 > Техника, страница 8 > Азотная кислота
> Техника, страница 8 > Азотная кислота
Азотная кислота
Азотная кислота, HN03, получается растворением окислов азота в воде:
3N02 + H20=2HN03 + N0
n2o3+h2o=hno3+no
N205 + H20-2HN03.
Физические свойства А. к. Мол. в.—63,016; бесцветная жидкость с характерным запахом; t°Kun. 86°, 1°пл.—47°; уд. в 1,52 при 15°; при перегонке благодаря разложению 2HN03=N203+20+H20 А. к. тотчас выделяет кислород, N203и воду; поглощение последней вызывает повышение t°nun. В водном растворе крепкая А. к. обычно содержит окислы азота, и приготовление совершенно безводной А. к. представляет значительные затруднения. Получить безводную А. к. перегонкой невозможно, так как минимум упругости имеют водные растворы А. к., то есть прибавление воды к кислоте и обратно понижает упругость пара (и повышает ί°κιιη.). Поэтому в результате перегонки слабой кислоты (_D<1,4) получается постоянно кипящий остаток D=1,415, с содержанием 68% HN03 и с t°Kun. 120°,5 (735 лш). Перегонка при пониженном давлении дает остаток с меньшим содержанием HN03, при повышенном давлении—с бблыпим содержанием HN03. Кислота Ό —1,503 (85%), очищенная продуванием воздуха от N204, дает при перегонке остаток с 77,1% HN03. Кислота D=l,55 (99,8%) дает при перегонке сначала сильно окрашенный оки азота раствор Ώ=1,62, а в остатке кислоту D=l,49. Т. о. в остатке при перегонке А. к. всегда оказывается кислота, соответствующая минимуму упругости (максимуму t°Kun.)· Безводную кислоту можно получить лишь при смешивании крепкой (99,1%) А. к. с азотным ангидридом. Вымораживанием, повидимому, нельзя получить кислоту свыше 99,5%. При новых способах (Валентинера) добывания А. к. из селитры, кислота получается достаточно чистой, при старых приходилось ее очищать преимущественно от хлористых соединений и от паров N204. Наиболее крепкая кислота имеет Лв=1,559, _D15=1,53, а 100%-ная HN03— Л4=1,5421 (Белей и Манлей); 100%-ная кислота дымит на воздухе и притягивает пары воды столь же сильно, как и серная кислота. Кислота с D=1,526 при смешивании со снегом нагревается.
Теплоты образования |из-*-Н2+ ^-N2+|-02j:
HN03 пар +34 400 cal
HN03 жидкость +41 600 »
HN03 кристаллы +42 200 »
HN03 раствор +48 800 » Теплоты разведения: при прибавлении к HN03 одной частицы Н2О — 3,30 Cal, двух частиц — 4,9 Cal, пяти частиц—6,7 Cal, десяти — 7,3 Cal. Дальнейшее прибавление дает ничтожное повышение теплового эффекта. В виде кристаллов получаются: 1) HN03.H20=H3N04— ромбические, напоминающие AgN03 таблички, ί°πΛ.=—34° (—38°); 2) HN03 (HaO)a=HeNOe — иглы, Ь°пл. —18°,2, устойчивы лишь ниже —15°. Кривая t° закристаллизовывапия водной кислоты имеет три эвтектики (при —66°,3, при —44°,2, при —43е) и два максимума (ΗΝ03.Η20 —38°, ΗΝ03·3Η20 —18°,2). Те же особенные точки наблюдаются для теплот растворения и для переломов кривой электропроводности, но на последней еще замечены 2ΗΝ03. Н20 и HN03. 10Н2О. Из только что сказанного и по аналогии с фосфорными к-тами следует, что в растворах А. к. имеется ее гидрат HN03, но он очень легко разлагается, что и обусловливает высокую реакционную способность HN03. А. к., содержащая в растворе Ν02, называется дымящей (красной).
Химические свойства. Чистая HN03 легко разлагается и окрашивается в желтоватый цвет благодаря реакции 2ΗΝ03=2Ν02 + 02 + Η20 и поглощению образовавшегося азотноватого ангидрида. Чистая А. к. и вообще крепкая А. к. устойчива лишь при низкой t°. Основным признаком А. к. является ее чрезвычайно сильная окислительная способность за счет отдачи кислорода. Так, при действии па металлы (кроме Pt, Rh, Ir, Au, на которые HN03 при отсутствии хлора не действует) А. к. окисляет металл с выделением окислов азота тем меньшей степени окисления, чем энергичнее в качестве восстановителя был окисляемый металл. Напр., свинец (РЬ) и олово (Sn) дают N204 [2HN03 + Pb=Pb0 + N204 + H20; Pb0+2HN03=Pb(N03)2 + H20]; серебро — преимущественно N203. Сера, особенно све-жеосажденная, окисляется легко, фосфор при легком подогревании превращается в фосфористую кислоту. Уголь, накаленный докрасна, загорается в парах А. к. и в самой А. к. Окисляющее действие дымящей красной кислоты больше, чем бесцветной. Железо, погруженное в нее, делается пассивным и уже не поддается действию кислоты. На циклические органические соединения (бензол, нафталин ит.п.) А. к. безводная или в смеси с серной к-той, действует очень сильно, давая нитросоединения С6Н5Н + HN03=CeHBNOa + НОН. Нитрация парафинов идет медленно, притом только при действии слабой кислоты (большая степень ионизации). В результате взаимодействия веществ, содержащих гидроксил (глицерин, клетчатка), с А. к. получаются азотнокислые эфиры,неправильно называемые нитроглицерином, нитроклетчаткой ит. п. Все опыты и всю работу с азотной кислотой необходимо вести в хорошо вентилируемом помещении, но лучше под специальной тягой.
Анализ. Для обнаружения следов А. к. применяют: 1) дифенилэнданилодиги-дротриазол (в продаже—«нитрон»); 5 или 6 капель 10 %-ного раствора нитрона в 5 %-ной уксусной кислоте приливают к 5—6 смг исследуемого раствора, прибавив к нему заранее одну каплю H2S04: в случае присутствия заметных количеств ионов N03 выделяется обильный осадок, при очень слабых растворах выделяются игольчатые кристаллы; при 0° можно открыть при помощи нитрона даже 80 q0q- ΗΝ03; 2) бруцин в водном растворе; смешивают с исследуемым раствором и осторожно приливают по стенке пробирки к крепкой серной кислоте; lia месте соприкосновения обоих слоев в пробирке образуется розовато-красное окрашивание, переходящее снизу в зеленоватое.
Для определения количества HN03 в растворе дымящей А. к. нужно протитровать N204 раствором КМп04, определить плотность жидкости ареометром и вычесть указанную в особой таблице поправку на содержание N204.
Промышленные способы добывания А. к. Добывается А. к. гл. обр. из селитры. Раньше добывание селитры велось вт. н. «селитряницах» (sal-petriere), или «буртах», где, в результате перемешивания навоза, мочи и тому подобное. со старой штукатуркой,постепенно, отчасти благодаря действию бактерий, происходит окисление мочевины и других органических соеди
Таблица уд. н. HNOs различной концентра-ц и η (по Л у н г е).
| Граду- | В 100 | В 1 | л (в кило- | |||||
| Vg | сы | весовых частях | граммах) | |||||
| « 1 | i Л
t=c К |
КИСЛО- | кисло- | |||||
| « Г" | m 4 | n,o5 | HNO, | ты | n2o; | HNOa | ты | |
| >> s | м | Η ё: | 4 0"Вё | 40° Вё | ||||
| 1,060 | 8,0 | 12 | 9,15 | 10.68 | 17,25 | 0,097 | 0,113 | 0,182 |
| 1,090 | 11,9 | 18 | 13,31 | 15,53 | 2 5,08 | 0,145 | 0,169 | 0,27 3 |
| 1,120 | 15,4 | 24 | 17,34 | 20,2 3 | 32,6 7 | 0,195 | 0,227 | 0,366 |
| 1,185 | 22,5 | 37 | 25,83 | 30,13 | 48,66 | 0,306 | 0,357 | 0,577 |
| 1,220 | 26,0 | 44 | 30,24 | 35,28 | 56,16 | 0,369 | 0,430 | 0,695 |
| 1,255 | 29,3 | 51 | 34,78 | 40,58 | 65,54 | 0,437
0,508 |
0,509 | 0,822 |
| 1,290 | 32,4 | 58 | 39, 39 | 45,95 | 74,21 | 0,593 | 0,957 | |
| 1, 320 | 35,0 | 64 | 4 3,47 | 50,71 | 81, 90 | 0,573 | 0,669 | 1,080 |
| 1,345 | 37,0 | 69 | 47.08 | 54,93 | 88,71 | 0,633 | 0,739 | 1 ,193 |
| 1,375 | 39,4 | 75 | 51,69 | 60,30 | 97,38 | 0,711 | 0,829 | 1,339 |
| 1,400 | 41,2 | 80 | 5 5,97 | 65,30 | 105,46 | 0,783 | 0,914 | 1,476 |
| 1,410 | 42,0 | 82 | 57,86 | 67,50 | 109,01 | 0,816 | 0,952 | 1,537 |
| 1,420 | 42,7 | 84 | 59,8 3 | 69,80 | 112,73 | 0,849 | 0,991 | 1,600 |
| 1.450 | 44,8 | 90 | 66,24 | 77,28 | 124,81 | 0,961 | 1,121 | 1,810 |
| 1, 500 | 48, 1 | 100 | 80,6 5 | 94,09 | 151,99 | 1,210 | 1,411 | 2,278 |
| 1 ,510 | 48,7 | 102 | 84,09 | 9 8,10 | 158,43 | 1,270 | 1,481 | 2,392 |
| 1. 520 | 49,4 | 104 | 8 5.44 | 99,67 | 160,97 | 1,299 | 1,515 | 2,447 |
ками (например Кубанская область), возможно наличие в почве заметного, но недостаточного для добывания, количества селитры. Заметные количества добывались в долине Ганга и находятся в наших средне-азиатских крепостях, где запасы содержащей селитру почвы доходят до 17 тонн в каждом месте, но содержание в ней селитры не больше 3%. Залежи натриевой селитры — чилийской— были открыты в 1809 г.; они находятся преимущественно в провинции Тарапака, между 68° 15 и 70° 18 в д. и 19° 17 и и 21° 18ю. ш., по встречаются и южнее и севернее (в Перу и в Боливии); месторождение их расположено на высоте 1100 метров над уровнем моря. Залежи имеют протяжение ок. 200 км дл., 3—5 км шир., содержание NaN03B среднем 30—40%. Запасов, принимая ежегодный рост потребления в 50 000 т, может хватить на 300 лет. В 1913 г. вывезено 2 738 000 т, но вывоз в Европу несколько уменьшился, хотя, после очень заметного падения вывоза во время войны, он снова несколько повысился с 1920 г. Обычно сверху лежит «костра» (50с.и — 2м толщ.), состоящая из кварцевого и полевошпатового песка, а под ней «калихе» (25 сантиметров — 1,5 л»), содержащая селитру (залежи находятся в пустыне рядом с залежами соли и борнокальциевой соли). Состав «калихе» очень разнообразен; в нем NaN03—от 30% до 70%, йодистых и йодноватых солей—до 2%, хлористого натрия—16—30%, сернокислых солей—до 10%, магниевых—до 6%. Лучшие сорта содержат в среднем: NaN03—50%, NaCl—26 %, Na2S04—6 %, MgS04—3 %. Растворение NaN03 ведется при высокой t°, чтобы в раствор перешло гораздо больше NaN03, чем NaCl, растворимость которого незначительно увеличивается с t°. Из 3 тонны «калихе» получается 1 тонна сырой селитры со средним содержанием 95— 96% селитры. Из 1 л маточного рассола обычно получается 2,5 — 5 г иода. Обычно сырая селитра бурого цвета, из-за примеси окиси железа. Для удобрения применяют селитру, содержащую до 1 — 2 % хлористых соединений. Чистый азотнокислый натрий бесцветен, прозрачен, не гигроскопичен, если не содержит хлористых соединений; кристаллизуется в кубах. Для получения А. к. селитру нагревают с серной кислотой; взаимодействие идет по ур-ию:
NaN03 + H2S04=HNOs + NaHS04,
т. e. получают кислый сульфат. Последний можно применить для добыванйя хлороводорода прокаливанием смеси NaHS04 и NaCl в муфелях. Для взаимодействия по уравнению нений азота (амины, амиды и тому подобное.) в А. к., образующую с известняком кальциевую селитру. В жаркие дни, особенно на юге (например в Индии и в Ср. Азии), процесс идет очень быстро. Во Франции в 1813 г. добывали из селитряниц до 2 000 000 килограмм селитры. 25 крупных животных дают около 500 килограмм селитры в год. В некоторых местностях, с основной почвой, богатой животными остат
2 NaNO. + H,S04=Na2S04 + 2HN03
170 “ 98 142 126
теоретически необходимо взять на 100 килограмм NaN03 57,6 килограмм H2S04 или 60 килограмм кислоты 66° В6. В действительности, во избежание разложения, серной кислоты берут на 20—30% больше. Взаимодействие ведут в горизонтальных цилиндрических железных ретортах 1,5 метров длины, 60 сантиметров диам., со стенками в 4 сантиметров толщ. В каждый цилиндр входит 75 килограмм селитры и 75 килограмм H2S04. Пары проводят сначала через керамиковый холодильник, охлаждаемый водой, или через наклонную керамиковую трубу, потом через поглотители: «баллоны» или «бонбоны», то есть большие керамиковые «вульфовы склянки». Если взята серная кислота 60° В6 (71%) и в первый поглотитель помещено 4 килограмма воды на 100 килограмм селитры, то получится кислота в 40 — 42° Be (38 — 41%); применив кислоту в 66° В6 (99,6%) и сухую селитру, получим 50° В6 (53%); для получения кислоты в 36° Be, в первый поглотитель помещают 8 л воды, во второй — 4 л, а в следующие по 2,6 л. Дымящую А. к. получают, действуя на селитру вдвое меньшим количеством серной кислоты, чем следует по расчету. По этому способу получается кислота, загрязненная хлористым нитрозилом и другими веществами, отходящими в начале процесса, и оки азота — в конце отгонки. Окислы азота сравнительно легко отогнать, продувая через кислоту ток воздуха. Гораздо выгоднее работа в ретортах, охватываемых со всех сторон огнем и имеющих снизу трубу для выпуска бисульфата, содержащего заметное количество кислоты. Дело в том, что чугун не разъедается кислотой, если он достаточно нагрет и если соприкосновение огнем со всех сторон гарантирует от осаждения капель кислоты. В подобных ретортах (1,20 μ шир. и 1,50 метров диам., со стенками в
4—5 сантиметров толщ.) селитру обрабатывают серной кислотой из расчета 450 килограмм и даже 610 килограмм селитры на 660 килограмм H2S04 (66° Be). Вместо баллонов теперь часто применяют вертикальные трубы или соединяют эти трубы с баллонами.
По способу Гутмана разложение производится в чугунных ретортах, составленных из нескольких частей (фигура 1 и 1а); части со
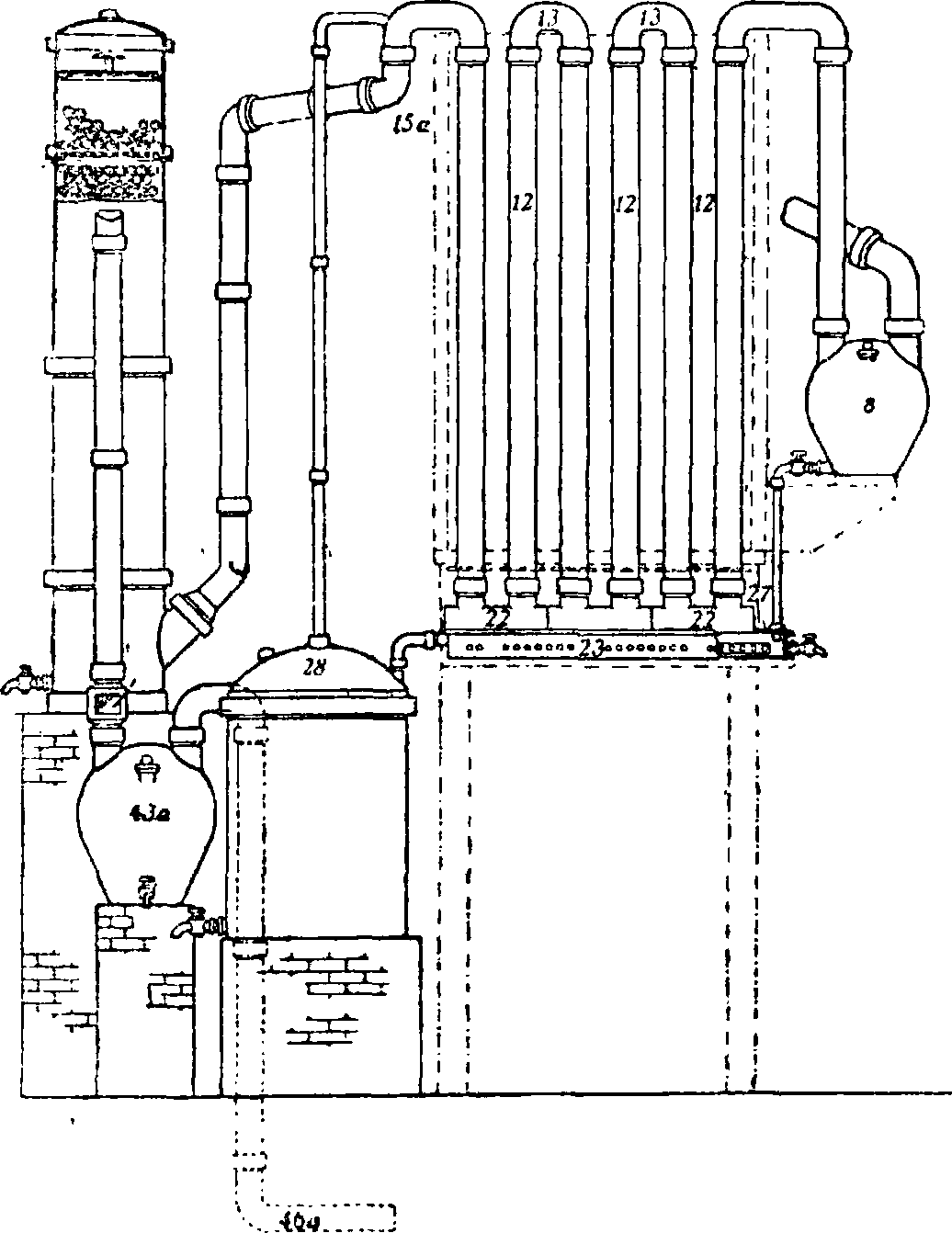
единены замазкой, состоящей обычно из 100 ч. железных опилок, 5 ч. серы, 5 ч. хлористого аммония с возможно малым количеством воды; реторты и, по возможности, загрузочный люк заключены в кирпичную кладку и нагреваются печными газами. В реторту загружают 800 килограмм селитры и 800 кв 95%-ной серной кислоты и ведут перегонку 12 час.; при этом тратится ок. 100 килограмм угля. Применяются также и цилиндрические реторты. Выделяющиеся пары попадают сначала в баллон 8; затем проходят ряд керамиковых труб, 12 и 13, помещенных в деревянный короб с водой; здесь пары сгущаются в А. к., которая сте- фщ-. lа. Чугунная реторта кает ПО трубам 22 установки Гутмана, и 23 в сборник 28,
сюда Hie попадает и конденсат из баллона 8; не сгустившаяся в трубах 12 А. к. попадает через 15а в башню, заполненную шарами и омываемую водой; последние следы кислоты, не поглощенные в башне, улавливаются в баллоне 43а", газы же через трубу 46а уносятся в дымовую трубу. Для окисления образующихся при перегонке окислов азота к газам непосредственно при выходе из реторты примешивается воздух. Если в производстве применяется крепкая серная кислота и высушенная селитра, то получается бесцветная 96—97%-ная А. к. Почти вся кислота конденсируется в трубах, лишь малая часть (5%) поглощается в башне, давая 70%-ную А. к., которую прибавляют к следующей загрузке селитры. Т. о. получается бесцветная А. к.,
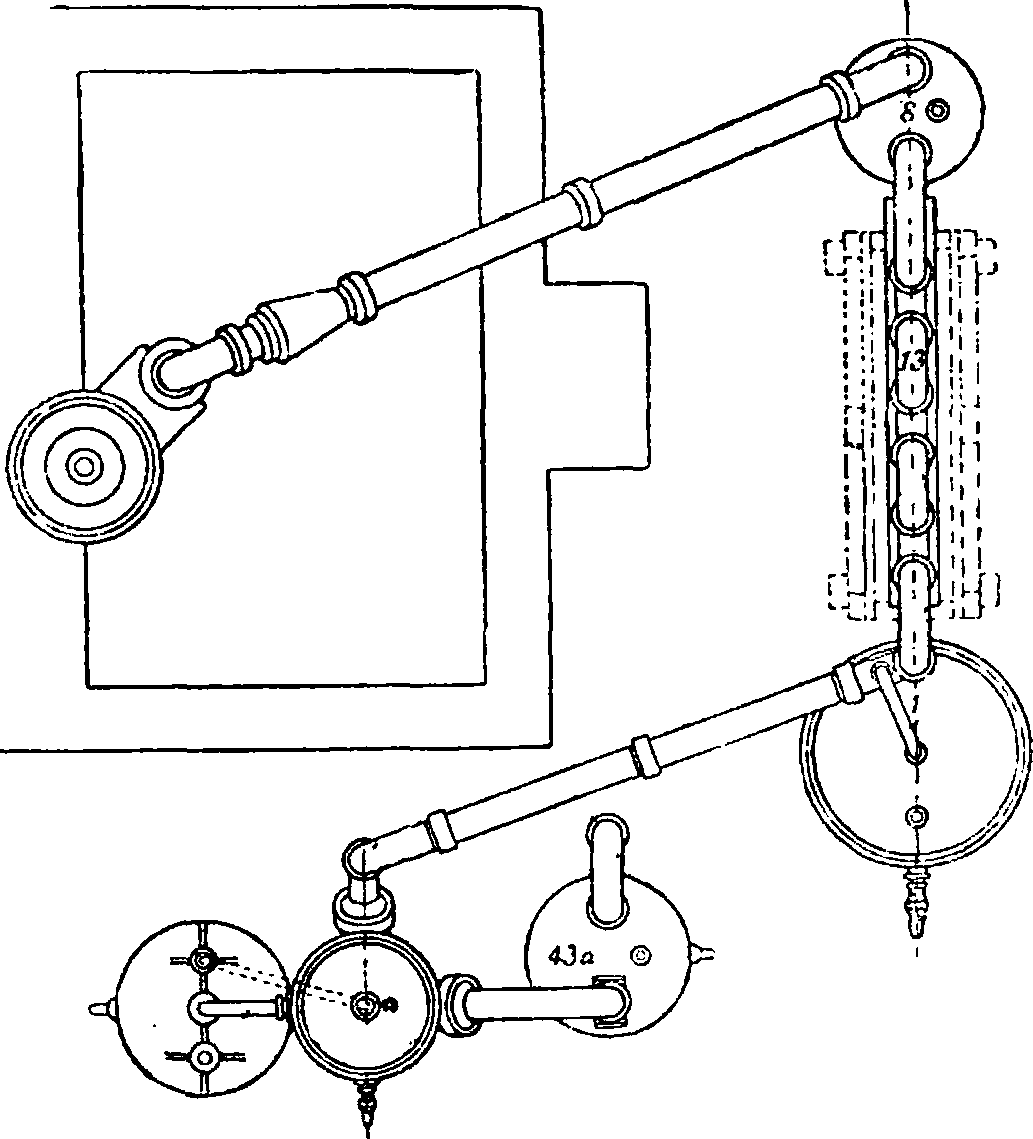
Фигура 1. Получение азотной кислоты из селитры (установка Гутмана). ·
лишенная хлора, с выходом в 98—99% от теории. Способ Гутмана получил большое распространение в виду простоты и дешевизны установки.
Из селитры добывают 96—100 %-ную кислоту по способу Валентинера, перегонкой под уменьшенным давлением (30 миллиметров) в чугунных ретортах смеси из 1 000 килограмм NaN03, 1 000 килограмм H2S04 (66°Ве) и такого количества слабой кислоты HN03, чтобы с ней ввести 100 килограмм воды. Перегонка идет 10 час., причем все время вводится воздух в сплав. Взаимодействие идет при 120°, но в конце процесса происходит «кризис» (1 час) и возможны сильные толчки (при 120—130°). После этого нагрев доводится до 175—210°. Весьма важно правильное сгущение и улавливание к-ты. Пары из реторты поступают в баллон, из него в 2 сильно охлаждаемых змеевика, из них в сборник (типа вульфовой склянки), за ним снова поставлен змеевик и дальше 15 баллонов, за которыми помещен насос. При 1 000 килограмм загрузки NaN03 в 6—8 ч. получается 600 килограмм HNO, (48° Вё), то есть 80% от нормы.
Для получения А. к. из норвежской селитры (кальциевой) последнюю растворяют, добавляют крепкую А. к. и примешивают серную кислоту, после чего отфильтровывают азотную кислоту от гипса.
Хранение и упаковка. Для хранения А. к. можно применять стеклянную, шамотовую и чисто алюминиевую (не больше 5% примесей) посуду, а также посуду из специальной кремнистой кислотоупорной стали Круппа (V2A). Т. к. при действии крепкой А. к. на дерево, опилки, тряпки, смоченные растительным маслом, и тому подобное. возможны вспышки и пожары (например, если лопнет бутыль при перевозке), то перевозить А. к. можно лишь в специальных поездах. Особенно легко при нагревании вспыхивает скипидар при попадании в крепкую А. к.
Применение: 1) в виде солей для удобрения, 2) для получения чатых веществ, 3) для получения полуфабрикатов для красящих веществ, а отчасти и самих красителей. Гл. обр. применяются соли А. к. или селитры (натриевая, аммонийная, кальциевая и калийная) для удобрений. В 1914 г. мировое потребление азота в виде чилийской селитры достигало 368 000 тонн и в виде А. к. из воздуха—10 000 ш. В 1925 г. потребление должно было дойти до 360 000 тонн А. к. из воздуха. Потребление А. к. сильно возрастает во время войны в виду траты на вещества, главными из которых являются нитроглицерин и нитроклетчатки разных типов, нитросоединения (нитро, тротил, мелинит и т. д.) и вещества для запалов (гремучая ртуть). В мирное время А. к. тратится на добывание нитросоединений, например нитробензола, для перехода к красителям через анилин, получающийся из нитробензола восстановлением. Значительное количество А. к. применяется для травления металлов; соли А. к. (селитры) применяются для чатых веществ (аммонийная селитра—в бездымных, калийная—в дымных ах) и для фейерверков (бариевая селитра—для зеленого цвета).
Стандарт азотной кисло-т ы. Стандарт А. к. существует пока только в СССР и утвержден Комитетом по стандартизации при СТО в качестве общесоюзного обязательного стандарта (ОСТ—47) для кислоты в 40° Вё. Стандарт устанавливает содержание HN03 в А. к. в 61,20% и ограничивает содержание примесей: серной кислоты не более 0,5%, хлора не более 0,8%, железа не более 0,01%, твердого остатка не более 0,9%; стандартная А. к. не должна содержать осадка. Стандарт регулирует взаимоотношения продавца и покупателя, жестко регламентируя методику отбора проб и производства анализов. Содержание А. к. определяется прибавлением к кислоте NaOH и обратным титрованием кислотой. Содержание серной кислоты определяется в виде BaS04 осаждением ВаС12. Содержание хлора определяют титрованием в щелочной среде азотнокислым серебром. Содержание железа определяют осаждением полуторных окислов аммиаком, восстановлением окисного железа в закисное и последующим титрованием КМп04. Упаковка А. к. не носит пока стандартного характера. Не касаясь размера, веса и качества тары, стандарт обусловливает упаковку А. к. в стеклянную посуду и дает указания, как ее упаковывать и закупоривать.
Получение азотной кислоты.
I. Из в оздуха. Синтез А. к. из воздуха при действии вольтовой дуги повторяет до известной степени процесс, совершающийся в природе под влиянием разрядов атмосферного электричества. Кавендиш первый наблюдал (в 1781 г.) образование окислов азота при горении Н2 в воздухе, а затем (в 1784 г.) и при проскаки-вании электрической искры через воздух. Мутман и Гофер в 1903 г. первые попытались изучить равновесие: N2+02$ 2NO. Пропуская через воздух вольтову дугу переменного тока в 2 000—4 000 V, они практически добились концентрации N0 от 3,6 до 6,7 объёмн. %. Расход энергии на 1 килограмм HN03 у них достигал 7,71 kWh. Это равновесие изучал затем Пернет, пропуская воздух через иридиевую трубку. Далее в том л<е направлении работали Нернст с Елли-неком и др. исследователи. Путем экстраполирования экспериментальных результатов исследования равновесия между воздухом и окисью азота Нернсту удалось вычислить, что в правой части ур-ия устанавливается при t° 3 750° (то есть приблизительно при t° вольт, дуги) содержание 7 объёмных % N0.
Приоритет идеи технического использования вольтовой дуги для фиксации атмосферного азота принадлежит фраиц. исследовательнице Лефебр, которая еще в 1859 г. запатентовала в Англии свой метод получения А. к. из воздуха. Но в то время стоимость электрич. энергии была слишком высока, чтобы метод Лефебр мог получить практическое значение. Следует указать еще на патенты Мак Дугаля (Ан. II. 4 633, 1899 г.) и на осуществленный в техническ. масштабе метод Bradley и Lovejoy, эксплоа-тировавшийся в 1902 г. американской фирмой Atmospheric Products С° (с 1 млн. долл, капитала) с использованием энергии
Ниагарского водопада. К этому же времени следует отнести попытки использования напряжения в 50 000 V1 для фиксации атмосферного азота, сделанные Ковальским и его сотрудником И. Мосьцицким. Но первый существенный успех в деле фабрикации азотной кислоты из воздуха принесла историческая идея норвежского инженера Биркеляида,которая заключалась в том, чтобы использовать для повышения выходов окис
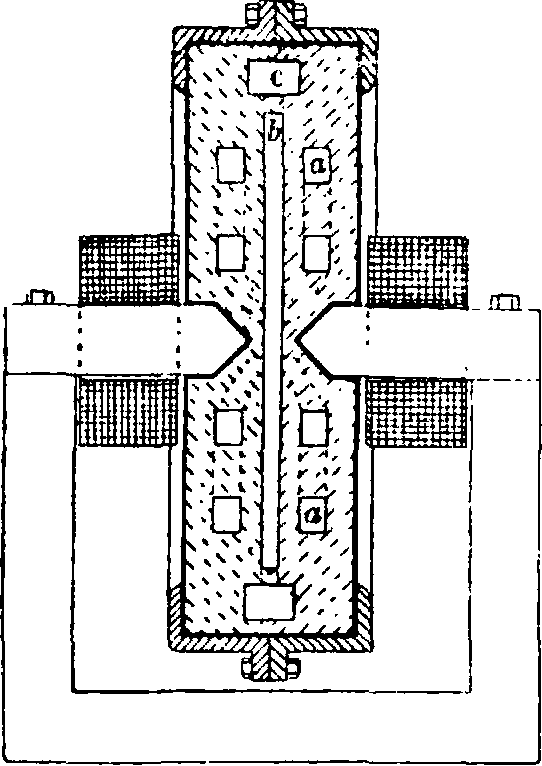
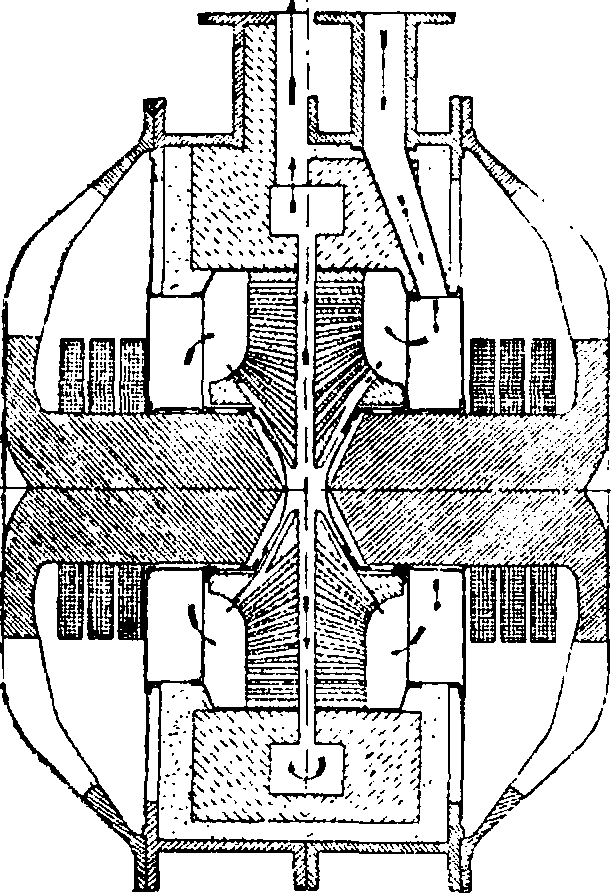
лов азота при пропускании через воздух вольтовой дуги способность последней растягиваться в сильном электромагнитном поле. Эту мысль Биркелянд со-Фигура и схема печи_ Бир- вместиос другим норвежским инж. Эйде претворил в технич. келянда и Эйде (первоначальная конструкция): а—каналы для притока воздуха, __ ь—камера для установку, сразу же вольтовой дуги, с выход ttorttivto пентябель-окнсленных газов. давшей рентаоель ную возможность получения из воздуха А. к. Благодаря постоянной перемене направления токаи действию электромагнита образующееся пламя вольтовой дуги имеет все время тенденцию как бы раздуваться в разные стороны, что приводит к образованию быстро перемещающейся все время со скоростью до 100 м/ск вольтовой дуги, создающей впечатление спокойно горящего широкого электрич. солнца диам. в 2 метров и более. Через это солнце непрерывно продувается сильная струя воздуха, а самое солнце заключено в окованную медью особую печь из огнеупорной глины (фигура 1,2 и 3). Полые электроды вольтовойду-ги изнутри охлаждаются водой. Воздух через каналы а в шамотовой кладке печи поступает в дуговую камеру b; через с окисленный газ покидает печь и охлаждается с использованием его тепла для нагревания кот-> лов выпарива-
Фигура 2. Схема печи Биркелян- тельных аппара-
да И ЭЙД%тр?вдпя™ВаЯ К0Н тов. После этого
N0 поступает в окислительные башни, где окисляется за счет кислорода воздуха до N02. Последний процесс является процессом экзотермическим (2NO-f 02=2N02 + 27Cal), и поэтому условия, увеличивающие поглощение тепла, значительно способствуют реакции в этом направлении. Далее, двуокись азота поглощается водой согласно следующим ур-иям:
3N02 + Н20=2HNO, + N0 и 2Ν02 + ΙΙ20=ΗΝ03 + ΓΙΝ02.
По другому способу, реагирующую смесь газов перед поглощением охлаждают ниже 150°; при этой t° обратное разложение— Ν02=Ν0 + 0 почти не имеет места. Имея в виду, что при некоторых условиях равновесие N0 + N02$N203 устанавливается с максимальным содержанием N203, можно получить, поливая горячие нитритные газы еще до полного их окисления, при t° от 200 до 300°, раствором соды или едкого натра, вместо азотнокислых солей—чистые нитриты (метод Norsk Hydro). При выходе из

Фигура 3. Большая печь па фабрике в Нотоддене (общий вид).
печи продуваемый воздух содержит от 1 до 2% окислов азота, которые сейчас же улавливаются встречными струями воды и затем нейтрализуются известью с образованием кальциевой, так называемым «норвежской» селитры. На проведение самого процесса N2 + Ojj^ 2NO—43,2 Cal требуется затрата сравнительно лишь незначительного количества электрич. энергии, а именно: для получения 1 тонна связанного азота в виде N0 лишь 0,205 kW-года; между тем в лучших современных установках приходится затрачивать в 36 раз больше, то есть ок. 7,3 и до 8 kW-лет на 1 ж. Другими словами, свыше 97% затрачиваемой энергии идет не на образование N0, а на создание для этого процесса благоприятных условий. Чтобы сдвинуть равновесие в сторону возможно большего содержания N0, необходимо пользоваться t° от 2 300 до 3 300° (содержание N0 при 2 300°—2 объёмных % и для 3 300°—6 объёмных %), но при таких t° 2 NO быстро распадается обратно на
N2 + 02. Поэтому в небольшую долю секунды необходимо удалить газ из горячих областей в более холодные и охладить его хотя бы до 1 500°, когда распад N0 протекает более медленно. Равновесие N2+02^2N0 устанавливается при 1 500° в 30 ч., при 2100° — в 5 ск., при 2 500° — в 0,01 ск. и при 2 900° — в 0,000035 ск.
Существенными усовершенствованиями по сравнению с методом Биркелянда и Эйде отличается метод Шбнгерра, сотрудника BASF. В этом методе, вместо пульсирующего и действующего все же с перебоями прерывистого пламени вольтовой дуги п е-ременного тока, применяется спокойное пламя сильного постоянного тока. Этим предотвращается весьма вредное для процесса частое задувание пламени. Такого же результата, впрочем, можно достигнуть и при вольтовой дуге переменного тока, но продувая воздух через сожигательное пламя не прямолинейно, а в виде вихревого ветра вдоль пламени вольтовой дуги. Поэтому печь м. б. сконструирована в виде довольно узкой металлической трубки, притом т. о., чтобы пламя дуги не кась ее стенок. Схема конструкции печи Шбнгерра изображена на фигуре 4.
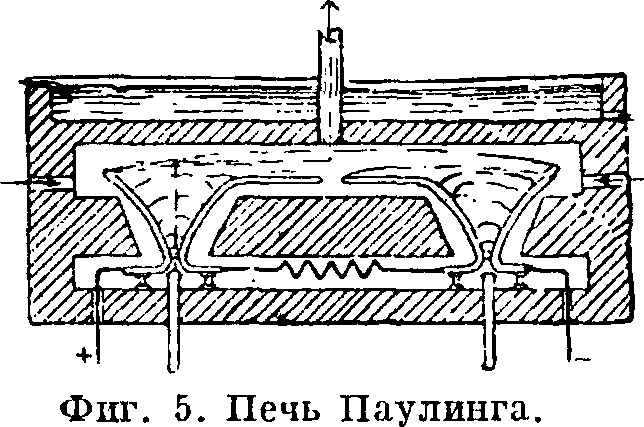
Дальнейшее усовершенствование в дуговой метод вносит метод Паулинга (фигура 5). Электроды в сожнгательной печи имеют вид роговых разрядников. Образующаяся между ними вольтова дуга в 1
1 метров длиной вздувается сильной струей воздуха, кверху. В наиболее узком месте оборвавшееся пламя дуга вновь зажигается при помощи дополнительных электродов.
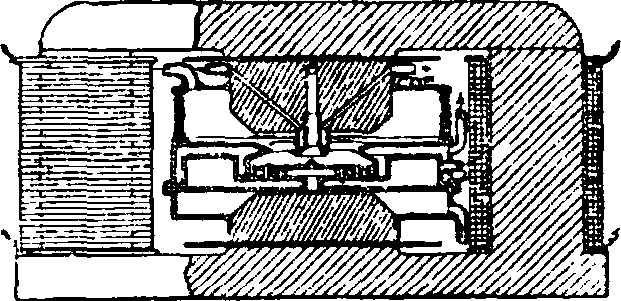
1]—Несколько иная конструкция печи для окисления азота воздуха запатентована И. Мось-цицким. Один из обоих электродов (фигура 6) имеет форму плоского диска и находится от другого электрода на весьма близком расстоянии.Верхний электрод трубчатый, и через него поступают быстрой струей нейтральные газы, распространяющиеся затем конусом. Пламя, вольтовой дуги приведено в круго
Фпг. 4. Печь Шбнгерра в разрезе: 1 —
сток воды, 2— приток воды, з— выход газов, 4 — приток воздуха, 5 — охлаждение электродов. вое движение под влиянием электромагнитного поля, а быстрая конусообразная струя газа препятствует коротким замыканиям. Подробное описание всей установки приведено у В. Waeser, Luftstickstoff-Indu-strie, р. 475, 1922. По методу И. Мосьциц-кого работает один з-д в Швейцарии (Chip-pis, Wallis), вырабатывая 40%-ную НМ03. Другой завод в Польше (Bory-Jaworzno)
рассчитан на 7 000 kW и должен вырабатывать концентрированную HN03h (NH4)2S04. Для улучшения выходов окислов азота и для повышения пламени вольтовой дуги, в последнее время применяется в качестве исходного продукта не воздух, а более богатая кислородом смесь азота и кислорода, с отношением 1 :1. С такой смесью работает французский завод в Ларош-де-Рам с очень хорошим результатом.
В настоящее время находятся в действии следующие предприятия, использующие дуговой метод для получения А. к.
| Место | Произв. способ, в т связ. N в год | Примечание |
| Рьюкан (Норвегия). | 28 000 | Работает полностью; |
| Ноттоден (Норвегия). | 7 000 | продукты — норв. и натрои. селитра, нитриты и конц. ΗΝΌ,
То же |
| Рина(Германия). | 4 000 | Не работает; продук- |
| Мульденштейн (Германия). | 2 000 | ты HNO, и нитрит То же |
| Пьерфитт (Франция). | 880 | Не работает; гл. про- |
| Ларош де - Рам (Франция). | 220 | дукт извести. селитра Раб.; гл. прод. HNO, |
| Пач (Австрия). | 1 000 | Работает; гл. продукт |
| Рим (Италия). | 500 | ΗΝΌ„ и нитриты Работ, с перебоями — |
| Ла-Гранд, Вашингтон (С.-А. | 270 | HNOs, NaNOs, KNO, и КСЮ,
Работаете перебоями; |
| С. Ш.). | гл. продукт NaNO, |
Получаемую четырехокись азота N204целесообразно сгущать в жидкость путем охлаждения до—90°. Такая жидкая четырехокись азота, полученная из предварительно высушенных газов — кислорода и воздуха, не реагирует с металлами и поэтому может транспортироваться в Стальных бом- Фигура 6. Печь Мосьцицкого. бах и служить для изготовления HN03 крепких концентраций. В качестве охлаждающей жидкости в этом случае одно время применялся, но, вследствие неизбежного просачивания окислов азота и действия их на, на з-дах Tschernewitz (в Германии) и Bodio (в Швейцарии) случились страшные ы, разрушившие оба предприятия. Извлечение N204 из газовой смеси м. б. достигнуто также при помощи абсорбции N204 силикагелем, выделяющим при нагревании поглощенный N204 обратно.
II. Контактным окислением аммиака. Все описанные методы получения синтетической А. к. непосредственно из
воздуха, как уже было указано, рентабельны лишь при наличии дешевой гидроэлектр. энергии. Проблема связан, азота (смотрите Азот) не могла бы считаться окончательно разрешенной, если бы не был найден способ получения сравнительно дешевой синтетической А. к. Усвоение связ. азота удобрений растениями особенно облегчено, если эти удобрения представляют собою соли А. к. Аммонийные соединения, внесенные в почву, должны предварительно подвергнуться нитрификации в самой почве (смотрите Азотные удобрения). Кроме того, А. к., наравне с серной кислотой, является основой многочисленных отраслей хим. промышленности и военного дела. Получение чатых веществ и бездымного а (тротил, нитроглицерин, пикриновая кислота и мн. др.), анилиновых красок, целлулоида и искусственного шелка, многих медикаментов и т. д. невозможно без А. к. Поэтому-то в Германии, отрезанной во время мировой войны блокадой от источника чилийской селитры и в то же время не располагавшей дешевой гидроэлектрич. энергией, в значительной степени развилось производство синтетической А. к. по контактному методу, исходя из каменноугольного или синтетического аммиака путем окисления его кислородом воздуха при участии катализаторов. Во время войны (1918 год) в Германии производилось до 1 000 тонн азотной кислоты и азотнокислого аммония в день.
Еще в 1788 г. Мильнером в Кэмбридже была установлена возможность окисления NH3 в окислы азота при действии перекиси марганца при нагревании. Кульман в 1839 г. установил контактное действие платины при окислении аммиака воздухом. Технически же метод окисления аммиака до А. к. был разработан Оствальдом и Брауэром и запатентован ими в 1902 г. (Интересно, что в Германии заявка Оствальда была отклонена в виду признания приоритета за франц. химиком Кульманом.) При действии мелкораздробленной платины и медленном течении газовой смеси, окисление идет по реакции 4NH3-f ЗОа=2N2 + 6H20. Поэтому процесс должен быть строго регулирован как в смысле значительной скорости движения газовой струи, продуваемой через контактный «конвертор», так и в смысле состава газовой смеси. Поступающая в «конверторы» смесь газов должен быть предварительно тщательно очищена от пыли и примесей, которые могли бы «отравить» платиновый катализатор.
Можно предполагать, что присутствие платины вызывает распад молекулы ΝΗ3 п образование нестойкого промежуточного соединения платины с водородом. При этом азот in statu nascendi подвергается окислению кислородом воздуха. Окисление NH3до HN03 протекает по следующим реакциям:
4NH3+5 02=4NO + 6 Н20;
охлажденный бесцветный газ N0, будучи смешан с новой порцией воздуха, самопроизвольно окисляется дальше с образованием N02 или Ν204:
2NO + Oa=2NOa, пли N8Ot;
растворение образовавшихся газов в воде в присутствии избытка воздуха или кислорода связано с дальнейшим окислением по реакции:
2N0a + 0 + H80=2HN0„
после чего получается HN03, крепостью примерно от 40 до 50%. Путем перегонки полученной HN03 с крепкой серной к-той можно получить, наконец, концентрированную синтетическую А. к. По Оствальду, катализатор должен состоять из мета л л ич. платины, покрытой частью или вполне губчатой платиной или платиновой чернью.
Реакция должна протекать при едва начавшемся красном калении п при значительной скорости течения газовой смеси, состоящей из 10 и более частей воздуха на 1 ч. NH3.
Медленное течение газовой смеси способствует полному распаду NH3 до элементов.
При платиновой контактной сетке в 2 сантиметров скорость течения газа должен быть 1—5 м/ск,г.е. время соприкосновения газа с платиной не должно превышать Vioo ск. Оптимальные t° лежат около 300°. Смесь газа предварительно нагревается. Чем больше скорость течения газовой смеси, тем больше и выход N0. Работая с применением очень густой платиновой сетки (катализатора) со смесью аммиака с воздухом, содерлчащей ок. 6,3% NH3, Нейман и Розе получили при t° 450° следующие результаты (при контактной поверхности пла-
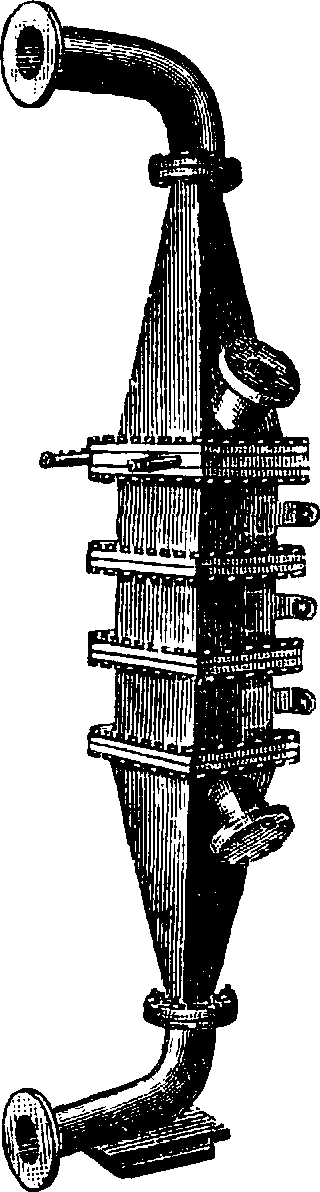
Фигура 7. Конвертор с платанов. сеткой.
| тины в 3,35 см2). Скор. теч. | Скор. теч. | Выход N1 |
| воздуха в лч | NН3 в л;ч | в % |
| 7,80 | 0,633 | 62,69 |
| 8,86 | 0,6 89 0,891 | 64,51 |
| 11,30 | 7 6,36 | |
| 16,86 | 1,050 | 91,22 |
| 19,80 | 1,267 | 96,06 |
| 23,09 | 1,738 | 9 5,80 |
| 26,98 | 1, 721 | 93,10 |
| 31,62 | 1,991 | 90,40 |
| Большее или : | меньшее | содержание |
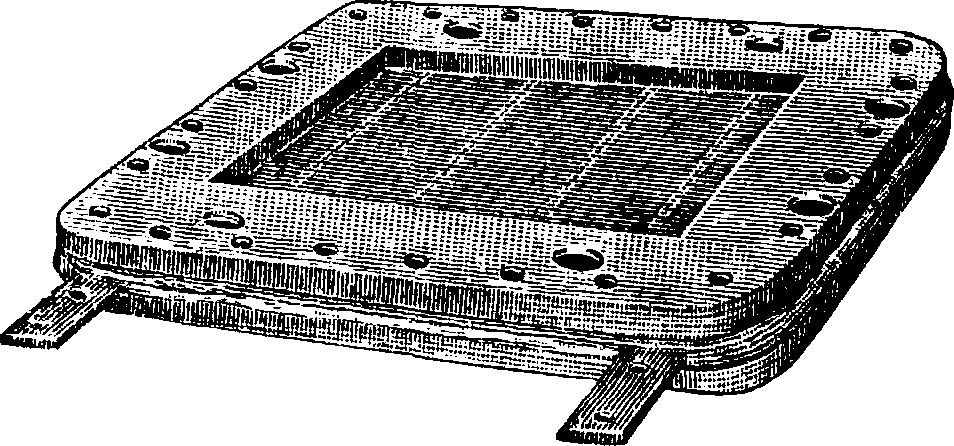
Фигура 8. Двойная платиновая контактная сетка нз конвертора. вления хим. процесса, который может идти или по ур-ию: 4NH3 + 502=4NO + 6Н20 (при содержании 14,38% NH3), или по ур-ию: 4ΝΗ3 + 702=4Ν02 +6Ы20 (при содержании в смеси 10,74% NH3). С меньшим успехом, чем платина, м. б. применены и другие катализаторы (окись железа, висмута, церия, тория, хрома, ванадия, меди). Из них внимания заслуживает только применение окиси железа при t° 700—800°, с выходом от 80 до 85% NH3.
Значительную роль при окислительном процессе перехода NH3 в HN03 играет t°. Самая реакция окисления аммиака экзотер-
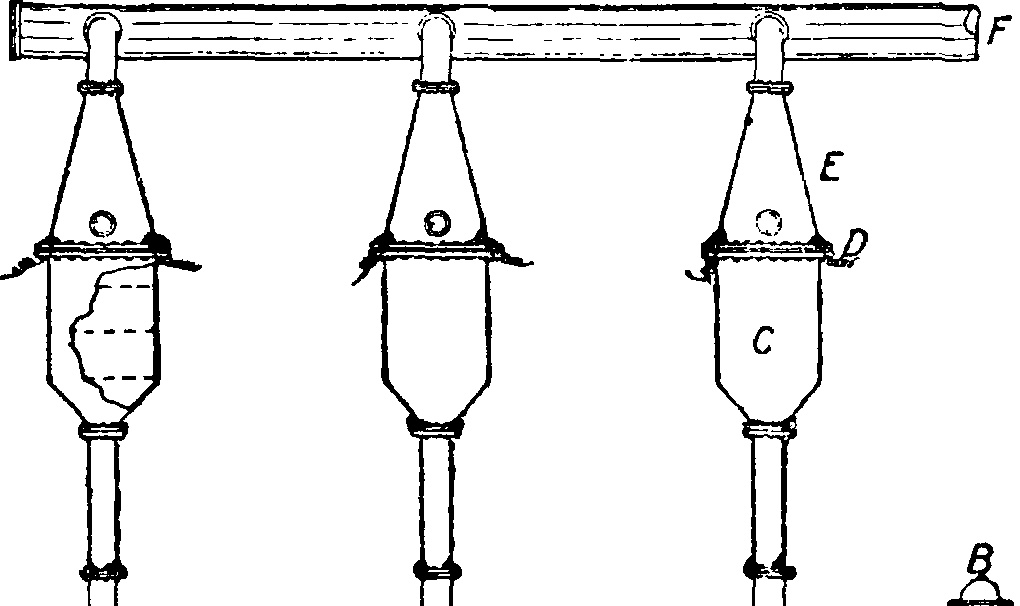
Фигура 9. Схема «конверторов» Франка-Каро: А—нагнетательный насос, В—газорегулятор, С—контактный аппарат, D—платиновая газовая сетка, нагреваемая электричеством, Е—алюминиевый шлем с слюдяным глазком, F—выход окислов азота. мична: 4ΝΗ,+50,=4Ν0 + 6Η,0+235,6 Cal.
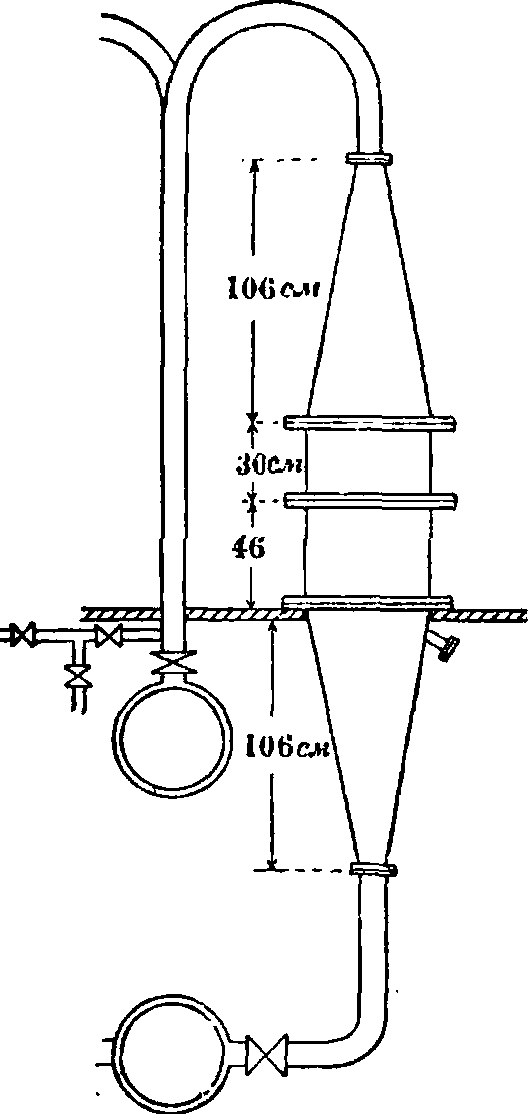
Лишь первоначально необходимо подогреть контактный аппарат,—далее реакция идет за счет собственной теплоты. Тех-нич. конструкция «конверторов» для окисления аммиака разных систем понятна из приведенных рисунков (фигура 7—8). Схема производства HN03по принятому в настоящее время методу Франка - Каро приведена на фигуре 9.
На фигуре 10 представлена схема окисления NH3 на фабрике Мейстера Люциуса и Брюниинга в Гехсте.
В современных установках окисление NH3 до N0 осуществляется с выходом до 90%, а последующее окисление и поглощение образовавшихся окислов азота водой—с выходом до 95%. Т. о., весь процесс дает выход связанного азота в 85—90%. Получение HN03 из селитры обходится в настоящее время (в пересчете на 100%-ную HN03)b 103 долл, за 1 т, по дуговому процессу 97 долл. 30 цент, за 1 т, в то время как 1 тонна HN03, полученной огсислением NH3, обходится всего 85 долл. 80 цент. Само собою разумеется, что эти цифры м. б. только примерными и в значительной степени зависят от величины предприятия, стоимости электрич. энергии и сырья, но все же они показывают, что контактному методу получения
HN03 суждено занять в ближайшем будущем господствующее положение сравнительно с остальными методами.
Лит.: см. Азот и Аммиак; Ullmann Fr., Enzykl. d. technisch. Chemie, B. 11; Waeser B., Die Luftslickstoff-Industrie, Lpz., 1922.