 > Техника, страница 95 > Азотная кислота
> Техника, страница 95 > Азотная кислота
Азотная кислота
Азотная кислота. Физические и химические свойства А. к. см. Азотная кислота, т. I.
Производство А. к. Основными источниками для производства А. к. являются аммиак, атмосферный воздух ц ископаемые, нитраты (чилийская селитра). Использование селитры потеряло присущий ему когда-то характер гегемонии и идет на убыль. Среди новейших изысканий по азотным проблемам весьма многообещающим является синтез А. к. из воздуха в высокочастотном разряде. Эта задача, являющаяся задачей энергетического выхода, еще ждет своего разрешения, но в случае успеха целесообразными окажутся такие процессы, как гидрирование окиси азота с целью получения аммиака или параллельный синтез А. к. и аммиака из элементов.
Еще несколько лет назад единственным источником производства А. к. в большом масштабе была натриевая селитра NaN03 (смотрите Азотная кислота, т. I). На смену естественному нитрату были выдвинуты методы фиксации азота jb элек-трич. дуге. Однако это производство захирело, и сейчас даже в Норвегии, стране с весьма дешевой гидроэнергией, заводы окисления азота кислородом воздуха перестроены почти целиком в з-ды синтеза аммиака. Причина заключается в высоком расходе энергии на реакцию
N2 + 02=2ΝΌ — 43 200 Cal, к-рая, будучи весьма эндотермической даже при г° вольтовой дуги, дает лишь 2—3% выхода N0 из воздуха. В тихих разрядах высокой частоты ионизация и активизация молекул приводят к реакциям типа:
N2 + 20=&ΝΌ + 100 000 Cal Л
2N + 20=2NO + 250 000 Cal;
t° реакции снижается до нескольких сот градусов, а выход возрастает до 10% и выше. Условия этих реакций еще изучаются и в промышленности не применены. Война 1914—18 гг. поставила государства перед лицом необходимости эмансипации производства А. к. Мобилизация изобретательской мысли привела к разработке и развитью метода окисления аммиака в окись азота с последующим превращением в А. к. Химизм этого процесса м. б. изображен ур-ием 4Ш13 + 502=4NO + 6Н20 + 216 700 Cal;
он был известен еще в 1800 г. (Фуркруа) и изучался в 1839 г. Кюльманом, впервые получившим А. к. из аммиака. Но до войны 1914—18 гг. во всем мире были построены лишь две небольшие установки, работавшие по способу Оствальда. В способе Оствальда было дано в простом виде основное техниц. решение проблемы; дальнейшие усовершенствования касались изменения состава катализатора, давления и t° отдельных стадий производства, на схема процесса осталась неизменной:
NH3 -»* NO -» N02 — N204 -> ΗΝ03.
Исходные вещества — аммиак, воздух, вода. В действительности помимо обратимого характера всех реакций при этом процессе переплетаются главные реакции с побочными, ведущими к образованию отбросных или первичных продуктов (N2, NO), и вследствие этого подбор оптимума времени конверсии, t°, давления теоретически довольно сложен. Интересно отметить, что в настоящее время термин «синтетическая А. к.» прилагается исключительно к кислоте, полученной окислением аммиака, тогда как продукт, полученный путем фиксации азота в вольтовой дуге, называется «дуговой» к-той. Уд. в трех основных способов получения А. к. — дугового процесса, окисления аммиака, разложения селитры — весьма различен; способ окисления аммиака асимптотически завоевывает 100%. Расход энергии на 4 тонн 100%-ной ΗΝ03 в т условного топлива: в дуговом процессе 14,2 тонны (ок. 18 000 kWh), синтез аммиака (0,7—1,1) плюс окисление аммиака (0,35 — 0,08) плюс концентрация кислоты (0,1 — 0,15)=1,0—1,5 тонн (ок. 1 25Э kWh). Этапы производства синтетич. А. к. таковы: 1) собственно окисление аммиака, или конверсия, 2) превращение N0 в N02, 3) растворение N02или его полимера N204 в воде с образованием А. к., 4) абсорбция хвостовых газов, 5) концентра ция полученной кислоты до необходимой крепости. В соответствии со специфичностью операций на з-дах обычно выделяют 1-й и 5-й этапы в самостоятельные цехи, а 2-й, 3-й, 4-й объединяют.
Окисление аммиака. В цехе конверсии происходит сжигание аммиака и приготовление смеси аммиака с воздухом или кислородом. Для технич. скорости реакции сжигание аммиака требует катализатора. Для этой цели изучались железные и платиновые контакты как в чистом виде, так и с добавками: к железу — марганца, меди, тория, висмута и других, к платине — родия, иридия. Неудобства железных катализаторов коренятся в небольшом диапазоне t°, в к-ром они обеспечивают достаточный % превращения NH3, хотя например Ре дает 90% контактирования NH3 при t° ок. 650° и в сравнительно малых объёмных скоростях (время контактирования в несколько десятков раз больше, чем на платиновом катализаторе). В настоящее время лишь один завод в Германии (Оппау) продолжает вповидимому работать на активированной окиси железа. Платина применяется либо в сплаве с 10% родия либо без него. Наиболее распространенная форма платинового катализатора — сетка с числом отверстий 1 000 — 3 600 на 1 см2 и толщиной нити — 0,04 — 0,09 миллиметров кое-где применяется фольга (способ УДЭ). Сетки обычно делают плоскими, но есть системы с ци-линдрич. сетками, соединяющимися не платиновым (Ni) дном. Применение сеток и установка их в виде цилиндра объясняется желанием развить большую поверхность соприкосновения с газами в данном объёме. Применение фольги исходит из иных целей: уменьшить потери платины вследствие уноса ее с газовой смесью; присутствие родия, не улучшая химич. качеств платины, также увеличивает ее долговечность. Действующая поверхность сеток на 1 см2 для плетения с нитью 0 0,065 миллиметров и 1 024 отв./см2, составляет 1,43 см2, для плетения с нитью 0 0,080 миллиметров и 3 600 отв ./см2 — 2,06 см2. Вес 1 м2 сетки равен в первом случае 525 г, во втором — 580 г. Потери платины- составляют на 1 тонна 100%-ной HN03для фольги 40 мг, для сетки при атмосферном давлении 70 мг, для сетки при давлении 7—8 atm 250 + 400 мг (не учитывая улавливания).
Роль давления при сжигании аммиака на платине показана на фигуре 1. Давление в цехе кон
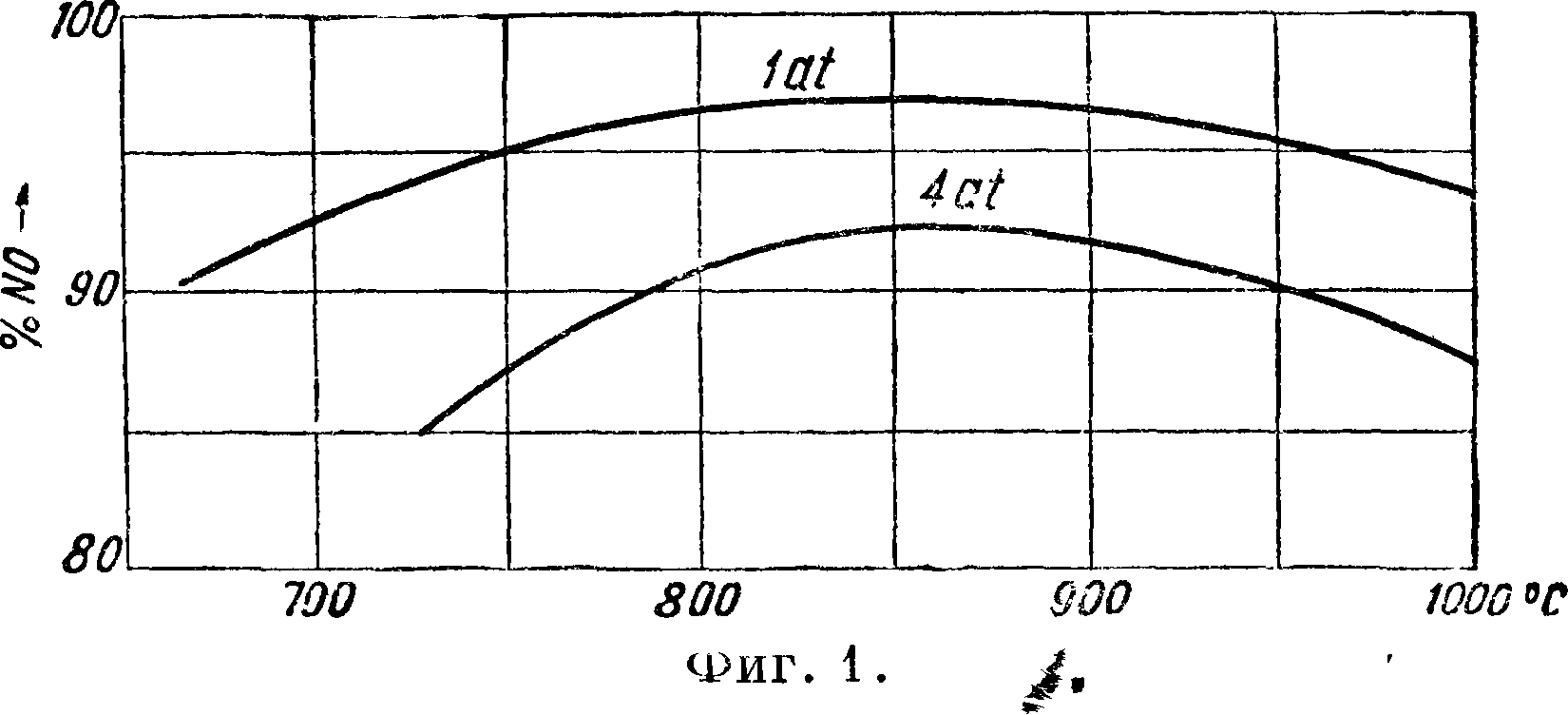
версии применяется не вследствие улучшения выходов окисления, а вследствие сравнительно небольшого их снижения при давлениях порядка 5—8 atm польза давления очевидна лишь для процессов окисления N0 в N02 и абсорбции; поскольку же технически до последнего времени было затруднительно изготовлять безупречные компрессоры для сжатия смеси после конвертера (т. к. они должен быть либо кислотоупорными либо должны работать при высокой ί°), то и было предложено подавать аммиак и воздух в контактный аппарат уже предварительно сжатыми. Дозировка аммиака и кислорода имеет огромное зна чение для ведения процесса. Во-первых, это влияет на реакционные объёмы (надо сказать, что до последнего времени в виду сравнительной дороговизны чистого кислорода и даже обогащенного воздуха в качестве окислителя применялся воздух), во-вторых, аммиак образует с воздухом и кислородом смеси при определенных соотношениях. Границы чатости: сухих аммиачно-воздушных смесей 16,1— 26,5% при 20°, 14.,0 — 30,4% при 250° и ходе газа вверх; сухих аммиачно-кислородных смесей 15,3 — 79,0% при 20° и распространении пламени вверх. Повышение давления до 10 atm понижает нижний предел чатости на 1 — 1,5%. Очевидно, что работать с той дозировкой кислорода, которая целесообразна для осуществления суммарной реакции
NH3 + 202=1ШОз + н20
(часть ее протекает в конверсии, конечные фазы— в абсорбции), в обычных условиях нельзя. Лишь за последние годы появились опытные полузаводские установки, разрешившие задачу сжигания аммиака в чистом кислороде с целью непосредственного (без концентрации) получения крепкой к-ты. Так, Зедерберг разработал схему, в которой контактная зона помещается под слоем охлаждающей жидкости — азотной * к-ты. Присутствие водяных паров действует антидето-нирующе; в этом же направлении влияет конструкция контактной плиты с капиллярами. Значительная часть абсорбции происходит в слое циркулирующей кислоты в конвертере, часть абсорбции осуществляется в специальных отдельно стоящих абсорберах. Процесс идет спокойно при 25—28% NH3 в смеси с 98% 02. Существуют предложения пропускать через конвертер совместно NH3, кислород и водяной пар. Надо сказать, что имеется возможность избежать концентрации и другим путем: посредством выделения N204 после конверсии в жидком виде, например конденсируя воду без давления, а первую порцию кислоты уже под давлением. Дозировка воздуха или кислорода одновременно с обеспечением объёмных соотношений в конверсии и абсорбции является средством регулировки t° в зоне сжигания аммиака. Упрощая химич. процессы при контактировании, удобно полагать, что аммиак сгорает в N0 по ур-ию
4NH3 + 502=4NO + 6Н20 + 216 700 Cal,
а побочные реакции можно свести к ур-ию 4NH3 + 302=2Ν2 -μ 6Н20 -ь 303 ООО Cal.
В действительности явление осложняется диссоциацией аммиака на элементы (к-рая однако протекает в ничтожной степени при том времени соприкосновения с катализатором, к-рое применяется в технике окисления NH3), диссоциацией N0 в зоне контакта (практически незаметна по тем же причинам, что и диссоциация аммиака) и наличием реакции
4ΝΗ3 -μ 6ΝΟ=5Ν2 -μ 6Н20 + 493 000 Cal.
Практически удается вести процесс конверсии с выходом N0 в 90 — 95% по объёму от аммиака. Остальная часть аммиака теряется вследствие окисления его до молекулярного азота. Ряд исследователей разрабатывал различные схемы истинного процесса сжигания NH 3, причем в качестве промежуточных продуктов реакции предполагались N2H2 (Рашиг), HNO (Андрусов), но ни одно из этих соединений обнаружено не было. Повышение f за счет теплового эффекта реакций над катализатором достигает 700°, при содержании 9% NH3 в аммиачно-воздушной смеси, что обеспечивает количество кислорода, необходимое для превращения N0 в к-ту (то есть NH3 : 02=1 : 2). Такая f достаточна для реакции, но повышение ее позволяет применять большие скорости газа (фигура 2, кривые одинаковой скорости газа)
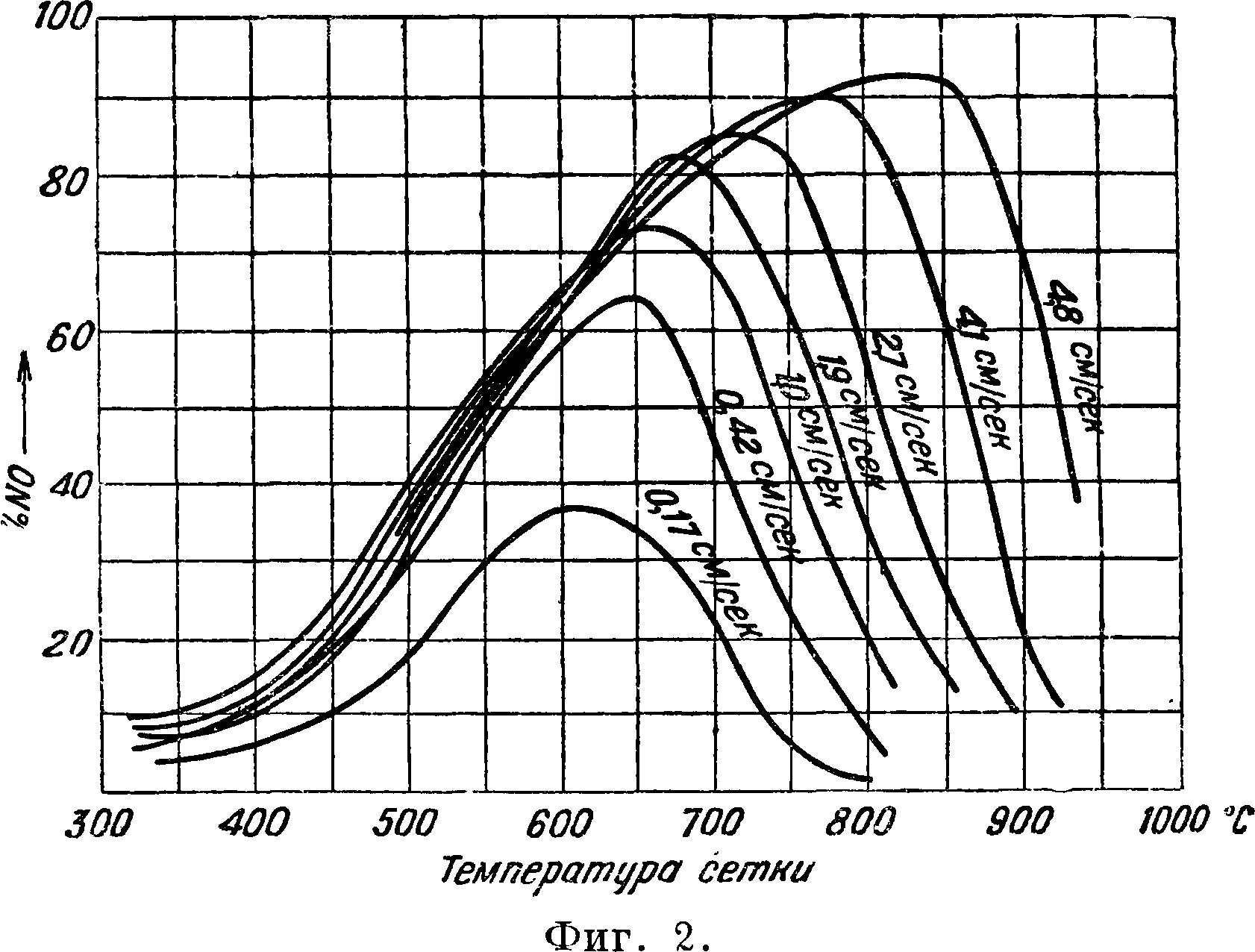
и этим достигать большей производительности контактных аппаратов. Замечательно, что с повышением скорости газа уменьшается влияние побочных реакций (диссоциации NH3 и взаимодействия NH3 с N0). В результате современная техника остановилась на скоростях в 20—10 раз больших, чем применявшиеся еще несколько лет назад. Классические исследования Неймана и Розе дали для t° 500° оптимальное время соприкосновения 8 · 10“4 ск.; Парсонс, работая при 900°, нашел 1 · 10’4 ск. и Пэрли для 1 025° — 0,5 · 10"4 ск. Явление удовлетворительно выражается ф-лой
Т=—282 lgZ,
где Т — темп-pa реакции и. Z — время соприкосновения. Один кг платиновой сетки сжигает 70—100 килограммjnac NH3 при t° 600° (о загрузке сеток можно судить например по производительности агрегата БАМАГ около 3,5 m/сутки NH3); 1 килограмм сетки сжигает 80—120 килограмм)час NH3 при t° 1 000° (под атмосферным давлением, производительность агрегата Парсонса — 2 mjсутки), в зависимости от формы плетения; повышение давления позволяет при неизменном времени соприкосновения увеличить количество пропускаемого газа; под давлением например 7 atm 1 килограмм сетки при 900 — 1 000° сжигает до 250—350 килограмм /час NH3 (производительность агрегата Дюпона — 4 — б иг/сутки). Необходимая t° м. б. обеспечена 1) предварительным нагревом газов, 2) обогащением смеси кислородом, 3) нагреванием зоны контакта. Последний способ неудобен, т. к. сокращает срок службы катализатора и вызывает расход энергии.
При работе без добавок кислорода обычно производится подогрев или воздуха или аммиачно-воздушной смеси за счет тепла газов, покидающих конвертер (иногда после использования части их тепла для обогрева дымогарного парового котла). Пуск конвертера производится водородным пламенем, овым факелом, специа^^юй электрической спиралью. Коммуникация, через которую проходит горячий аммиак, не должна содержать металлов кроме алюминия и никеля; прочие вызывают диссоциацию NH3. Конвертер выполняется в виде цилиндра или двух конусов, сложенных основаниями, с алюминиевыми (для t° околЬ* 600° и ниже) или футерованными стенками. Ход газа в целях сокращения оопасного диапазона обычно устраивается снизу, вверх, нижняя часть конвертера имеет насадку для рекуперации тепла (например керамиковый бой). При наличии на комбинате дешевого кислорода, например при соседстве с цехами ректификации воздуха, применяется дозировка кислорода как средство регулировки t° например в количестве ок. 4% в смеси (рабочая t° 800°, Фаузер). Такое количество кислорода в смеси практически не отражается на концентрации кислоты, и следовательно 02 здесь применяется исключительно как средство увязки теплового баланса. Газовая смесь составляется путем смешения газообразных аммиака и воздуха, прошедших фильтры, или например продувкой воздуха через аммиачный раствор; в последнем случае дозировка проводится изменением t° раствора и давления воздуха. При конверсии под давлением аммиак можно испарять при условиях, обеспечивающих давление аммиака не ниже, чем в системе (соответствующая t° пара); воздух сжимают отдельно. Чистота газовой смеси имеет огромное значение: 1) присутствие в газе 0,38% С2Н2 или 0,02% H2S или 0,00002% РН3 вместо 94% выхода N0 от NH3 дает через 8 ч. выход 70%. Смазочное масло, попадая на контакт, действует, как С2Н2, но слабее. 2) Железо, попадая на платину, аннулирует в этом месте ее каталитич. действие. Платина, улетучиваясь с контакта, оседает на стенках конвертера в виде платиновой черни и увеличивает % образования азота. Все эти явления, равно как и постепенное понижение активности платины при работе с чистым газом, приводят к необходимости ее смены или регенерации после 2 000— 4 000 час. работы без давления; при работе под давлением срок службы без регенерации уменьшается почти в 10 раз. Регенерация производится промывкой в крепкой соляной к-те (из раствора выделяют платиновый шламм, который удается улавливать в размере ~ 4% от веса уноса). Изменение состава газа в конвертере иллюстрируется следующим примером (в %):
NH3 02 N н20 N0
До контакта. 9,5 38,9 71,6 -- —
После контакта. — 7,06 71,0 13,69 8,25
Переработка окиси азота в А. к. Основные реакции таковы:
I. 2N0 + 02=2N02 + 27 000 Cal
И. 2N02=N204 + 13 600 Cal
III. Ν02 + Ν0=Ν203 + 10 310 Cal
IV. 2N02 + H20=HNOs-aq + HN02-aq + 27 730 Cal’
V. N203 + H20=2HN02 · aq + 13 300 Cal’
VI. 3HN02=HN03 · aq + 2NO + H20 - 18 130 Cal[
Как видно из сопоставления (IV) и (VI) реакций, 1/3 N02 вновь превращается в N0 параллельно с образованием А. к. Кроме того азотистый ангидрид N203 также выделяет N0 [после протекания (V) и (VI) реакций] в количестве, составляющем а/3 исходного объёма. Т. о. в условиях абсорбции должен быть обеспечено непрерывное при различных концентрациях в газовой фазе окисление N0; окисление N0 и растворение N02требуют б. или м. продолжительного времени. Поскольку N02 и N204 образуют с водой А. к. и азотистую к-ту согласно правой части ур-ия (IV) лишь с различным тепловым эффектом, процесс полимеризации двуокиси азота в четырех-окись особенного внимания не привлекает. Можно отметить, что при г° выше 60° и парциальном давлении NO а вплоть до 0,1 atm степень полимеризации не превышает 0,1; при этом же давлении при г° 0° степень полимеризации достигает уже 0,8. Степень полимеризации N02 следует учитывать при различных тепловых расчетах. N203образуется лишь при недостаточном окислении
N0 в N02 в соответствии с оставшимся количеством окиси азота (NO+N02=NaOs). Понижение t° благоприятствует всему циклу абсорбции: реакции (I—V) сдвигаются направо, а (VI) налево. Но замечательно, что и скорость окисления N0 в N02 тоже благоприятно изменяется, увеличиваясь с понижением t° (в химии известно лишь несколько подобных примеров). Темп-рный коэф. К скорости реакции, отражающий изменение скорости при увеличении t° на 10°, для темп-рного интервала 0—10° равен 0,912:
К =
wt + ю wt
0,912.
С повышением температуры температурный коэф. увеличивается, приближаясь к единице, и например для t°=300° К=0,997. Данные константы равновесия реакции NO -> N02 позволяют вычислить для парциального давления N0 в 0,1 atm степень окисления: 0,05 для 670°, 0,95 для 230°. Практически при t° свыше 600° в газе оказываются лишь следы N02. Но кинетика реакции требует для достижения значительной степени окисления гораздо более низкой ί°, что указывается равновесным состоянием. Полагая реакцию тримолекулярной, имеем
^jj- — Ka2 (1—x)2(b—αχ),
где 2 a — начальная концентрация NO в моль /л, b — начальная концентрация 02, х — степень окисления в долях единицы, t — время окисления, a (1 — х) — половина молей NO, ах — половина молей N02, (b — ах) — концентрация кислорода. В результате получим
_ 2,303 Г х(b-а). J 1-ос
Кф-а)2 2,303а(1—х) ^ 19 ,_ах *
b _
Здесь К — f(t) — коэф. скорости прямой реакции (фигура 3, константа скорости реакции).
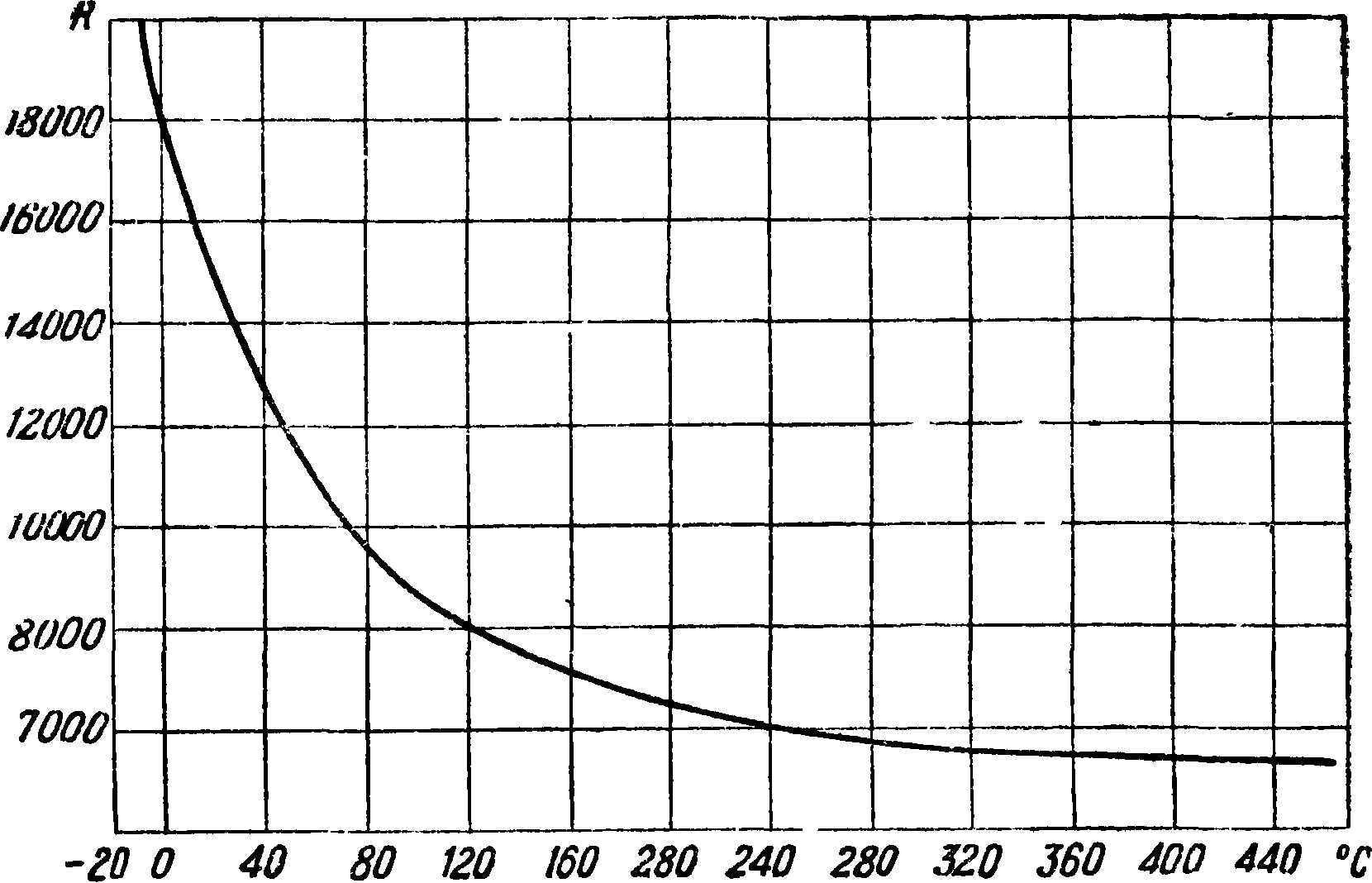
Фигура з.
Эта формула дает близкие к действительности цифры (табл. 1) и применима в пром-сти окисления S02 в S03 (камерным или башенным способом).
Таблица 1. — Время окисления N0 (в ск.) в зависимости от давления (ί° — 90° и N0 : 02=1:0,9).
| Степень окисления | |||
| PN0, aim | 0,8 | 0,9 | 0,95 |
| 0,09 | 97 | 258 | 601 |
| 0,45 | 4 | 11 | 24 |
Боденштейн полагает, что отрицательный температурный коэф. скорости объясняется меньшей вероятностью тройного удара молекул с повышением кинетич. энергии; Гаше и Патрик утверж-
дают, что реакция окисления N0 протекает в несколько фаз,-например через N204 и др., и соотношение скоростей отдельных различных реакций таково, что для более низких t° N0 образуется в конечном счете быстрее. В технич. практике малая скорость окисления N0 приводит к тому, что определенную долю газа оказывается невыгодным улавливать в виде А. к. Действительно, для средней t° в 20° зависимость объёма абсорбции (в м3 на 1 тонна аммиака в сутки) от степени абсорбции оказывается следующей:
% абсорбции. 95 93 90 85 80 70 60
Объем абсорбции. 200 130 100 70 60 50 40
Условия процесса: атмосферное давление, аммиачно-воздушная смесь. Повышение давления до 7 atm уменьшает объём реакционного пространства в 20—25 раз. Обогащение кислородом при N0 : 02=1 : 1,6 и 17% азота в газе (93%-ный кислород как источник 02) уменьшает объём в 3 раза. Пастонези сообщает, что пущен з-д, на к-ром окисление N0 ведут в присутствии силикагеля как катализатора. Повышение давления при абсорбции, помимо снижения капиталовложений, позволяет получать более концент-рированцую к-ту, т. к. соответственно сдвигается равновесие и увеличивается скорость реакции (IV). Системы под атмосферным давлением дают кислоту 40—50%-ную, под давлением 3—7 atm 55—62%-ную. Теоретич. предельная концентрация без отвода воды 77%; стремиться к ней нецелесообразно, т. к. с приближением к равновесию в реакциях (I) и (IV) необходимое время соприкосновения чрезвычайно увеличивается, и вместо секунд требуются часы. Степень достижения равновесия в реакции (IV) уменьшается с увеличением концентрации к-ты, уменьшением парциального давления окислов азота и увеличением скорости газового потока.
В результате реакции (IV) и (VI) имеем 3Ν02 + Н20=2HN03+ N0,
откуда
τη _ ρΝΟ · p2HN03р ρ3Ν02 · рН20 ’
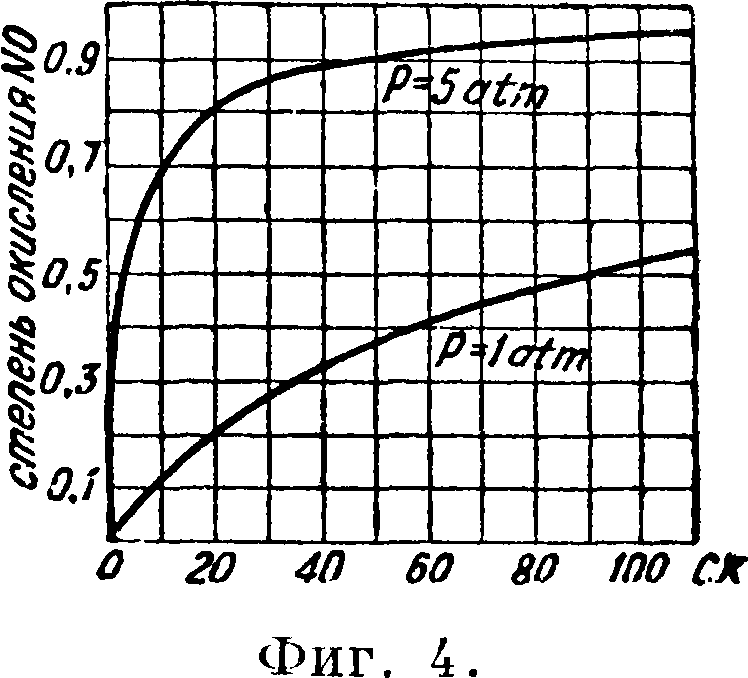
где Р — соответственные парциальные давления, а Кр — константа равновесия. Однако теоретич. подсчет не дает достаточно надежных цифр. Обычно предел водной абсорбции составляет 92—97% от N0 после конверсии, а остаток N0 поглощается вместе с Ν02 (в виде газа с 0,1 — 0,5% NO -f Ν02, 4 — 5% 02, прочее Na) в щелочах (едких, углекислых) или серной к-те, образуя соответственно нитрит-нитраты и нитрозу. Нитрит-нитраты подвергают инверсии продувкой воздухом и в виде нитрата калия или натрия после выпарки выпускаются как товары. Нитрозу наиболее предпочтительно использовать в качестве обезвоживающего компонента при концентрации слабой А. к. При малых количествах хвостовых газов изредка применяют известь в качестве поглотителя вследствие неприятных свойств ее суспензии. Интересна сравнительная оценка по-ттенш t° и повышения давления как средств увеличения скорости окисления N0 (фигура 4 и 5 — степень и время окисления): охлаждение газа с 30 до 0° уменьшает время окисления N0 (при Pno=0,1 atm, х — 0,9, ΣΡ=1,0 atm) с 300 до 140 ск.; эта затрата энергии при применении искусственного охлаждения, например аммиачных машин, эквивалентна сжатью до 1,8 atm; такое сжатие · сокращает время окисления до 80 ск. Следовательно компримирование примерно в 2 раза выгоднее охлаждения. Для характеристики состояния техники А. к. интересно отметить,
что еще недавно строились установки с абсорбцией под вакуумом на выходе вплоть до 250 миллиметров Hg. Предел давления определяется сравнительной стоимостью энергии и металла; работают системы от 2 до 8 atm абс. Предел охлаждения определяется наивысшей точкой плавления гидратов ΗΝ03, именно HN03 · ЗН20 с t°3ame. —18,5°.
Обычно охлаждение не ведут ниже —10°. Боль-
| 200 | |||||||||||||||
| 160
1?0 |
|||||||||||||||
500 600 700 800 900 ЮОО П001200
Секунд“
Фигура 5.
шинство установок интенсивного типа базируется на давлении, а не на охлаждении. Охлаждение водой конечно является одним из существенных элементов установки. Горячие нитрозные газы t° 1 000 — 600° охлаждаются в дымогарных трубках паровых котлов и затем с t° 500—300° поступают в теплообменники. Иногда устанавливают лишь одно из этих двух холодильных устройств. В водййых холодильниках г° газа опускается до 60—80°. Здесь получается нек-рое количество кислоты (5—10% в установках без давления, слабой; 30—60% в установках под давлением, крепкой); затем газ проходит или окислительные башни (полые пространства которых обеспечивают малые линейные скорости газа и следовательно предоставляют ему время для окисления) и поступает в абсорберы с насадкой для увеличения поверхности соприкосновения, орошаемые противотоком кислоты, или непосредственно поступает в абсорберы, вверху которых предусмотрено пространство для окисления N0, оставшегося от предыдущих стадий процесса и образовавшегося в самом абсорбере. Общий объём абсорберов определяется как величина, обратная времени окисления. Объем каждого отдельного абсорбера определяется конструктивными соображениями и выбором материала. А. к. требует применения ферросилиция или специальных хромовых (18—14% Сг) и хромоникелевых (8% Сг, 18% Ni) сталей или кисло-тоупора (керамики, гранита). Например для абсорберов применяется в США сталь: 15% Сг, 0,5% С, 1% Si, остальное Fe. Хромоникелевая сталь легче обрабатывается, но хромовая сталь дешевле. (Нитрозные газы с парами воды не корродируют обычно железо и сталь.) Абсорберы обычно устанавливают последовательно, с индивидуальными холодильниками, так как процесс поглощения N02 экзотермичен, а повышение t° нежелательно. Наибольшие неудобства гранита и андезита в том, что отвод тепла приходится производить лишь вне башен и в металлических холодильниках. Крупные отдельные абсорберы и башни из гранита выгодны.
Охлаждение м. б. или орошением снаружи (Фаузер), или посредством холодильников, включенных неоднократно по ходу газа и кислоты в абсорбере (Дюпон), или путем охлаждения вытекающей из абсорбера кислоты и выходящего газа (прочие системы). Повышение t° в абсорбере между холодильниками смотря по его размерам достигает 5—40°. Подобные схемы абсорбции в зависимости от исходных нитрозных газов или от ассортимента продукции можно значительно варьировать. Напр. отходящие газы установки денитрации серной к-ты, остаточные газы нитрования могут дать ценные азотсодержащие продукты в кислой или щелочной абсорбции. Устанавливая щелочное поглощение не в хвосте, а в начале системы, для горячих газов получаем почти чистый нитрит (520—670. г/м NaN02, 30 г/м NaN03), потребляемый на з-дах красителей и др. Наконец были попытки заменить водную абсорбцию поглощением аммиачной водой, однако при этом вместо реакций IV и VI текут реакции
VII. 2NOo + 2NH4OH=νη4·νο3 + νη4 · νο2 + н20 и
VIII. νη4νο2=Ν2 + 2Н20.
Последняя реакция приводит к тому, что выход в абсорбции снижается до 60—70%. Можно также комбинировать окисление N0 с другими производствами — серной к-ты, антрахинона; но эти процессы еще не вошли в производство.
Характеристики наиболее распространенных систем производства А. к. приведены в таблице 2. i
Расход энергии в таблице указан без учета рекуперации. Уже работает несколько з-дов, которые используют энергию расширения хвостовых газов в качестве привода; рекуперация достигает 40— 50%. Хвостовые газы перед рекуперацией подогреваются в теплообменнике до 230—270° для увеличения отдачи, нитрозные же газы в комбинированном способе, наоборот, следует охлаж дать для уменьшения объёма у всаса компрессора; но если желательно избежать применения специальной стали, вся компрессия должна вестись выше точки росы А. к., то есть выше 150°. С физико-химич. точки зрения наиболее совершенна схема, сочетающая конверсию без давления с абсорбцией под давлением, в металлич. сосудах с интенсивным охлаждением. Лишь недавно металлургия и машиностроение позволили добиться такой схемы; она является схемой с наибольшими шансами на распространение. При тщательном подборе условий конверсии и небольших давлениях с ней в некоторой степени может конкурировать процесс, подобный дюпоновскому, в особенности при организации улавливания уноса платины.
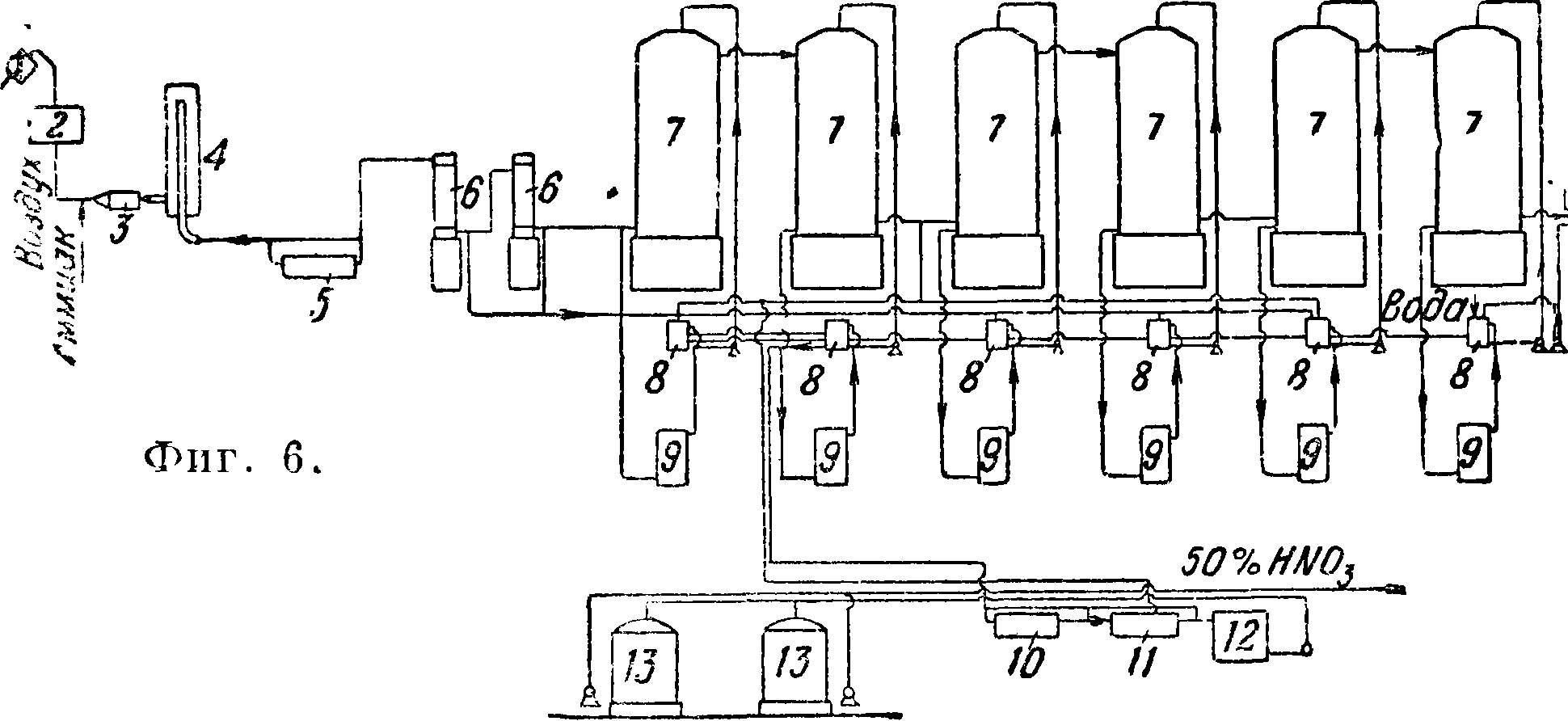
Как пример огромного абсорбера можно привести сконструированную фирмой УДЭ башню диам. 20 метров и высотой 20 м, разделенную внутри на секторы. Абсорберы бывают и с тарельчатой насадкой (Дюпон например диам. 1,5 м, высота 10 ж) и в виде лежачих цилиндров (Фаузер, диам. 3,5 ж, длина 12 ж). В системе м. б. один абсорбер (Дюпон) или ряд их в зависимости от давления и % абсорбции. Пример установки без давления— фигура 6: 1 и 2 — фильтры, 3 — смеситель, 4 — контактный аппарат, 5— паровой когел, 6 — газовые холодильники, 7 — абсорбционные гранитные башни, 8 — приемники, 9 — холодильники, 10 — подогреватель, 11 — аппарат для удаления азота, 12 — резервуар, 13 — хранилище готовой А. к.,
14 — щелочные башни,
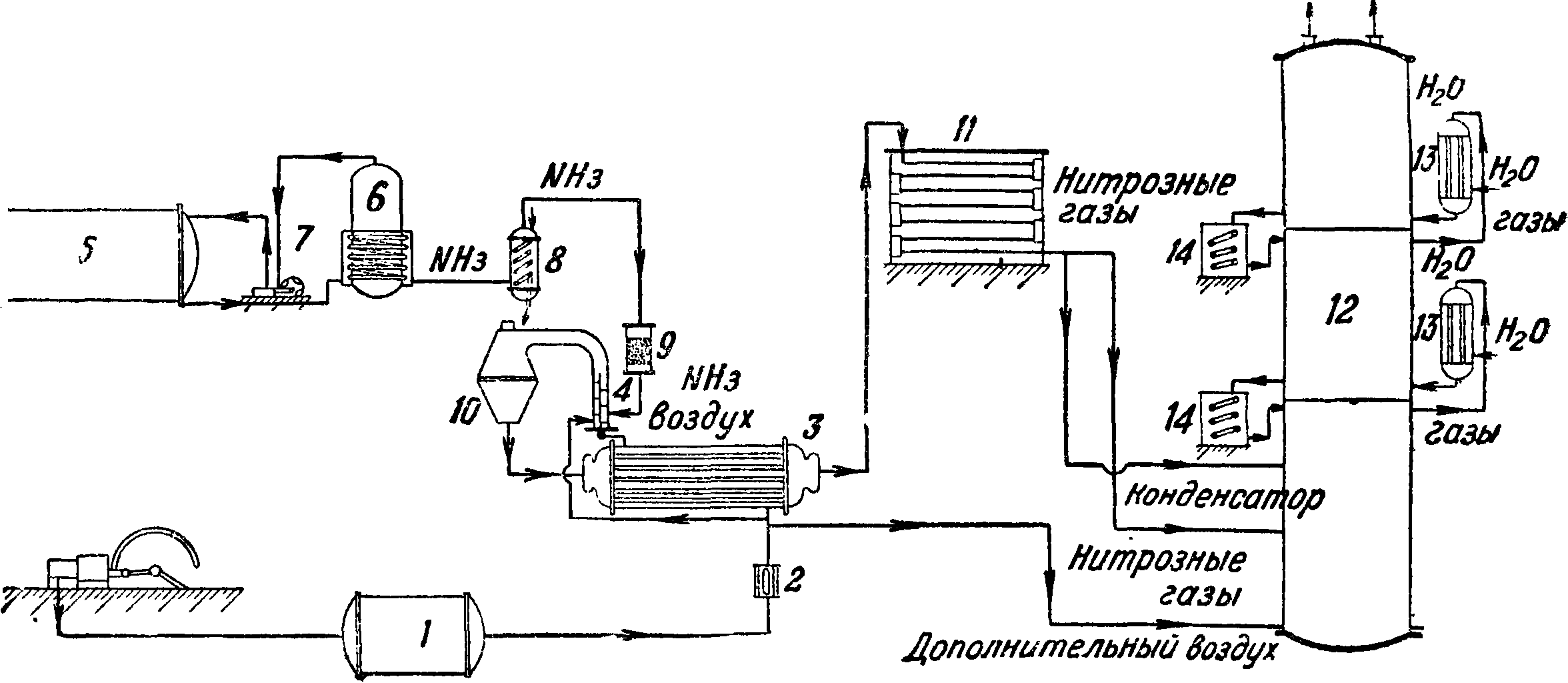
15 — сборники. Пример ус тановки под давлением— фигура 7: 1 — воздушный резервуар, 2 — воздушный фильтр, 3 — теплообменник, 4 — смеситель, 5 — хранилище для жидкого NH3, 6 — весовой танк, 7 — аммиачный насос, 8 — испаритель, 9 — аммиачный фильтр, 10 — конвертер, 11 — конденсатор, 12 — абсорбционная колонна, 13 — газовые холодильники, 14 — кислотные холодильники.
Концентрирование А. к. Все описанные методы дают к-ту, непригодную для ряда важнейших целей: для нитрации требуется гораздо более высоко концентрированная к-та. Как уже отмечалось выше, существуют способы окисления NH3 в кислороде в стадии опытной установки, и совсем недавно успешно начал работать завод Фаузера, дающий путем умелой комбинации основных факторов конверсии и абсорбции 98%-ную к-ту. В обычных условиях абсорбции N02 помимо реакции частично растворяется в к-те, которую даже приходится отбеливать продувкой воздухом; при этом часть N02 превращается в к-ту,
Таблица 2.—П р и мерные характеристики.
| Основная характеристик.i | Под давлением 7 atm | Конверсия без давления, абсорбция 4 aim | Без давления | |
| Характер катализатора | D=200 миллиметров *α Pt + 10% Rh 20—16 плоских сеток | 1400 миллиметров“2 Pt
2 плоские сетки |
Длина=30 миллиметров *з Ширина=10 миллиметров Толщина=0,02лш Pt
(фольга) |
D=1 500 лш“4 Pt
2 плоские сетки |
| Питание конвертера | Подогрев теплообме ном | 4% свободного кислорода | Подогрев теплообменом | Подогрев электричеством или теплообменом |
| 1 Темн-ра контакта | 950° | о о
00 | 700° 1 600° | |
| Производительность i агрегата в конверсии в т NH3 (в сутки) | 5 | 3 | 0,12 i 4 | |
| Выход в конверсии (в %) | 96 | 96 | I
94 1 96 | |
| Выход в кислой абсорбции (в %) | 98 | 98 | 92 | 94 |
| Расход анергии kWh на 1 m HN03 | 410 | 380 | 65 | 90 |
| Расход пара на 1 m
HN03 (в m) - |
0,6 | 0,9 | 0,6 | |
| Расход воды | 140-350 | 150 | 145 | 145 |
| Объем jvt3 абсорбции на 1 m NH3 (в сутки) | 4,5 | 30 | 125 | 125 |
| Щелочное поглощение (в %) | — | - | 7 | 4 |
| Материал абсорберов | Хромовая сталь | Никелъхро-мовая сталь | Гранит | Никельхро-мовая сталь |
| Крепость к-ты | 57—62 | 55-60 | 50 | 45-50 |
| Фирмы: *1 Дюпон. *2 Фаузер. *з УДЭ. *4 БАМАГ. | ||||
часть улетучивается. Медленно, под давлением может происходить реакция:
2N204 + 2Н20+02=4HN03 + 18 800 Cal.
Для обеспечения этих материальных соотношений конверсию проводят с воздухом под атмосферным давлением, -быстро охлаждают газ, чтобы дать минимуму N0 окислиться, и отводят конденсирующуюся воду в виде 3%-ной к-ты.
N Из газ
Затем остаточные газы сжимаются до 8—10 atm и направляются при t°
— 10° в абсорбер, орошаемый 60%-ной А. к. (отсюда газы идут на обычную абсорбцию). Раствор NOg в А. к. направляют в автоклав, куда вводят кислород, и при t° 70° под давлением 50 atm через 4 часа образуется 98%-ная к-та. Учитывая рекуперацию энергии, Фаузер сообщает, чго ему удалось добиться расхода энергии лишь в 130 kWh на 1 тонна HN03. Прежние попытки осуществить подобную схему вследствие низкого давления NOa е, нитрозных газах из аммиачно f воздушной смеси приводили к ^ выпадению его в твердом виде при вымораживании, что чрезвычайно осложняло работу, или требовали конверсии с чистым кислородом. Обычные схемы требуют концентрации кислоты в случае ее назначения в качестве нитрующего средства. А. к. с водой образует постоянно кипящие смеси с максимальным давлением; для атмосферного давления температура такой точки равна 121,9°; в смеси 68,4% HN03. При нагревании водных растворов азотной кислоты крепостью выше 68,4% с повышением точки кипения в газовой фазе происходит обеднение кислотой, что видно из табл. 3.
Таблица 3.— Процентное содержание HN03в водных растворах при температуре кипения. ляет 4,5—6,0 тонн при исходной концентрации 45— 50% HN03. В случае предварительной концентрации кислоты выпариванием до 60% расход серной кислоты уменьшается до 3,5 тонн При предварительной концентрации и применении глухого пара взамен острого в концентрационных колоннах расход серной кислоты можно свести до 1,75 тонн Интересно отметить, что в ряде случаев реторты для производства А. к. I ^ по способу Валентинера исполь-|{§ зуются как перегонные кубы для §|*» концентрации слабой синтетич. SjL к-ты. Если на комбинате отсут-jp-J ствует самостоятельное серно-JJI г-^сиш кислотное хозяйство, то уста-новке концентрации А. к. сопутствует установка реконцентрации серной к-ты. Применяются установки различных типов от широко известных аппаратов Кесслера до недавно появившихся барабанных концентраторов Ке-мико (смотрите Серная кислота) и установок концентрации под вакуумом.
Применение А. к. и статистические данные. Не являясь предметом широкого потребления, А. к. играет колоссальную роль в качестве сырья для производства удобрений,
| JO
1 пип.. |
85,5° | 112° | 121,9° | 112° | 106,5° |
| Жидкая фаза. Газовая фаза. | 96,0
99,9 |
80,0
97,0 |
68.4
68.4 |
33,0
5,9 |
24,2
2,16 |
Для производства кислоты крепостью выше 68,4% необходима перегонка с водуотнимающими средствами; обычно для этой цели применяется крепкая серная к-та. Процесс идет в дистилляцион-ных колоннах с тарельчатой или сплошной насадкой; вверху подается смесь для обработки; пары крепкой А. к. (95—96%) выходят также сверху, направляясь на конденсацию; внизу вытекает разбавленная во время операции до 73% серная к-та со следами А. к. Расход 96%-ной серной кислоты на 1 m 100%*ной HNOa состав-
бо%нт
Фигура 7.
чатых и красящих веществ. Статистич. данные по производству А. к. и связывания азота воздуха см. Аммиам.
Лит.: Эпштейн Д., Синтетическая азотная кислота,. М—Л., 1933; Торсуе в, «ЖХП», 1930,324; Либинсон, там же, 1931, 432; Маляревский, П а п к о в, там же, 1928, 682; Waeser W., Die Luft+ickstoffmdastrie, Lpz., 932; Curtis H., Fixed Nitrogen, N. Y., 1932; Pascal P., Syntheses et catalyses industrielles, P., 1924; Webb H., Absorption of Nitrous Oases, L. 1928; Taylor, «Ind. Eng. Chem.», 1931, p. 800: Fauser, «Chemical a. Metallurgical Engineering», N. Y., 1930, 604: 1928, 474; 1932, 430; Pastonesi, «Gnornale di chimica industriale ed applicata», Milano, 1933, 13; OCT
5374, 5375. Ю. Севастьянов.