 > Техника, страница 9 > Ализарин
> Техника, страница 9 > Ализарин
Ализарин
Ализарин, 1, 2-диоксиантрахинон СО ОН
0Н III! или С14Н804,
встречается в природе в виде глюкозида в зрелых корнях (krapp, garance, madder) т. н. красильной марены (Rubia tinctorum) и ее разновидностей. Мареновые — многолетние растения с лежачим стеблем; известны человеку как красильный материал с древнейших времен (Египет, Индия, Персия, Смирна). В Леванте корень марены носил название «лизари» или «ализари», отсюда—«ализарин». В Европе красильная марена разводилась гл. обр. во Франции (близ Авиньона), а также в Эльзасе, Бельгии, Голландии (с 16 в.) и Баварии. Со второй половины 18 века марена культивировалась в Крыму и на е (Дербент, Баку), а также в Самарканде. Ко времени открытия синтетического А., то есть к 70-м гг. прошлого столетия, мировая добыча маренового корня превышала 75 000 тонн (4 у2 млн. п.) — соответственно 750 тонн (45 000 — 50 000 п.) А., на сумму 75 000 000 фр. Общая площадь, занятая под культуры марены, исчислялась к этому времени тысячами км2. Цена ализарина составляла в то время приблизительно 100 франков за 1 килограмм.
В мареновом корне содержится глюко-зид А. в смеси с глюкозидом пурпурина (1, 2, 4-триоксиантрахинон), ксантопурпу-рина (1,3-диоксиантрахинон) и псевдопурпурина (3-карбоновая кислота пурпурина). Глюкозид А., т. н. рубэритриновая к-та, в чистом виде впервые получена Рохледером. Состав ее С26Н28014 установлен Грэбе и Либерманом. При нагревании с разведенными к-тами рубэритриновая к-та распадается на А. и глюкозу. Рубэритриновая к-та одноосновна; содержащаяся в ней биоза переходит при гидролизе в 2 молекулы глюкозы: С14Нв02(0Н) · (ОС12Н21О10) + 2НаО -> С14Н602 (ОН)2 + 2С6Н1206. Естественным путем расщепление глюкозида марены происходит под влиянием содержащегося в самом корне азотистого энзима, носящего название «эритрозима». Техническое выделение ализарина, более или менее свободного от пурпурина и других красящих спутников водных вытяжек маренового корня, разработано Коппом. В основе метода лежит подмеченное Коппом свойство глюкозида А. расщепляться труднее сравнительно с глюкозидами пурпурина и псевдопурпурина. Подкисленной сернистой к-той водой из свеже - измельченного маренового корня удается извлечь все красящие начала, не опасаясь потерь от преждевременного расщепления глюкозидов. При прибавлении 2 — 3% соляной кислоты и повышении t° до 60° выделяются красные хлопья пурпурина, в смеси с псевдопурпурином. Глюкозид А. количественно остается в растворе и м. б. выделен дальнейшим нагреванием в виде так называемым «зеленого А.». Впервые А. был выделен из краппа Робике (Robiquet) и Колэном (Colin) в 1826 г. Пурпурин выделен ими же в 1828 г. Выяснению строения А. способствовало наблюдение Шунка, получившего в 1848 г. при окислении А. к-ту, названную им ализариновой к-той, соединение, к-рое, по исследованиям Гебгардта, а затем Вольфа и Штрекера, оказалось тождественным с фталевой к-той. Вследствие этого А. стал считаться тогда производным нафталина, и ему приписывали формулу С10НвО3. В 1866 г. Штре-кер предлагает для А. формулу С14Н804, но на это сообщение в то время не было обращено должного внимания. Лишь Грэбе в 1868 г., применив к А. классический метод Байера—перегонку с цинковой пылью, показал, что родоначальником А. является антрацен. Уже в январе 1869 г. Грэбе и Либерман делают знаменательное сообщение о синтезе А., исходя из антрацена, и обращают внимание на научное и техническое значение этого открытия как первого примера искусственного получения растительного красителя. Исходным продуктом для этой работы послужил англ, каменноугольный антрацен, предоставленный Мартиусом в количестве 500 г. Первоначально Грэбе и Либерман при синтезе А. исходили из дибромантрахинона, сплавлением последнего с едким кали, затем—из антрацена рядом операций: антрацен ди-бромантрацен -» тетрабромид тетрабром-антрацен дибромантрахинон ализарин. Синтез А. (1869 г.) в связи с быстрым технич. усовершенствованием синтетических методов свел в течение немногих лет культивирование красильной марены нанет, освободив громадные пространства плодородной земли под другие культуры. Т. о. не только научное значение (первый синтез естественного красителя), но и практическое значение синтеза А. по своим экономическим последствиям является в истории беспримерным.
Современная нам формула строения А. создалась не сразу. Из двух возможных формул строения антрацена
сн сн
I.
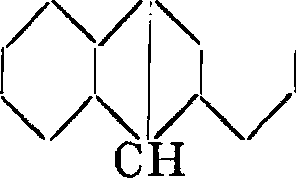
и II.
сн
ч/ч/ч
ч/
Грэбе и Либерман вначале склонны были отдавать предпочтение формуле II. Дальнейшие исследования Фиттиха и Остермайера и Грэбе вскоре показали, однако, что формула II как нельзя лучше отвечает строению фенантрена. Для антрацена оставалась т. о. лишь формула I. Фиттихом же было установлено, что антрахинону следует придать строение дифенилен-дикетона
СвН4<
свн4.
При окислении А. образуется незамещенная фталевая кислота свн4
/СО
Чсо/
С6
Н2(ОН)2-*С6Н
/СООН
4 соон’
значит оба гидроксила молекулы расположены в одном и том же боковом ядре. Наличие двух гидроксильных групп явствует между прочим и из того обстоятельства, что, как показал Шютценберже, А. образует дибензоилпроизводное. Работы Гримма, Байэраи Каро, Либермана и др. устанавливают относительные положения гидроксильных групп в А., гистазарине, хинизарине и пурпурине. Строение хинизарина как 1,4-диоксиантрахииона явствует из синтеза этого соединения конденсацией фталевого ангидрида с гидрохиноном. Конденсация фталевого ангидрида с пирокатехином протекает с образованием двух изомерных диоксиантрахинонов А. наряду с гиста-зарином:
г. /С0
/ч/со П.| I
0+ ί
он
он
со он
/Ч/Ч/Чон
21
ч/
С(
СО
0-L
он
/и
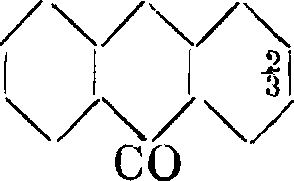
он он
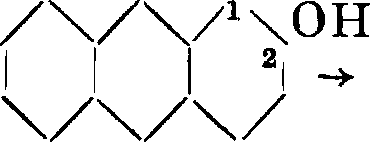
В соединении I одна из гидроксильных групп находится в положении 1, другая— в положении 2 по отношению к среднему ядру. Гидроксилы изомера II занимают положение 2 и 3. Мы знаем, что как 1-окси-, так и 2-оксиантрахинон при сплавлении со щелочами переходят в один и тот же А.
со он
/Ч/ч/ч

со он со
/Ч/Ч/ЧОН /ч/Ч/ЧОН
•II I 21 I I I 21 ч/ч/ч со со
Т. о. в А. гидроксильные группы должны занимать положение 1,2. Строение пурпурина как 1,2,4-триоксиантрахинона вытекает из того обстоятельства, что как А., так и хинизарин при окислении переходят в один и тот же пурпурин
СО ОН
со
СО ОН
п он
*1. ί
со он
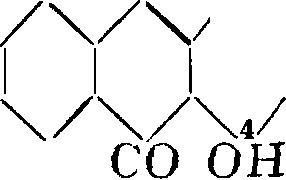
. ч со он
Строение антрапурпурина и флавопурпу-рина обусловливается строением исходных дисульфокислот антрахинона:
СО СО ОН
S0,H/4/4/4S03H ОН/Ч/ЧЧЧОН
‘ 17 Г I 21
ч/ч/ч со
(антра- или изопурпурин)
СО со он
/4/4/4S0.H /Ч/Ч/ЧОН
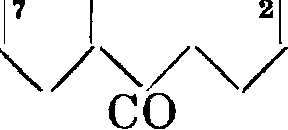
! 21
S08H4/4/4
со онч/ч/ч со
(флавопурпурин)
Метод Грэбе и Либермана синтеза А. из дибромантра-хинона. Метод этот был запатентован в Англии, Франции, Пруссии, России и Америке. Задачу выработки фабричного продукта взяла на себя Баденская ф-ка, которой прежде всего пришлось преодолеть трудности в получении сырья (антрацена). Каро первый показал, что при нагревании антрахинона с крепкой серной к-той до 200° образуется сульфокислота, переходящая при сплавлении с едким кали в А. Почти одновременно открытие это было сделано и в Англии Перкином (W. Н. Perkin). Каро, Грэбе и Либерманом метод этот был заявлен в Англии к патенту 25 июня 1869 г., Перкином—на один день позже. В отличие от Каро, Грэбе и Либермана, получивших в главной массе моносульфокислоту антрахинона, Перкин получил дисульфокислоты, дающие при сплавлении со щелочью А. более желтого оттенка. Патенты были выданы на обе заявки. Независимо, осенью 1869 г., тот же способ получения А. был найден на ф-ке бр. Гессерт в Эльберфельде химиком Борном и, еще несколько ранее (апрель 1869 г.), химиком Ризером на фабрике М. Люциуе-Брюннинг. В прусском патенте на этот метод Грэбе и Либерману было отказано, в виду того, что в их методе не было признано каких-либо существенных нововведений. Замена сульфо-группы на гидроксил сплавлением ароматических сульфопроиз-водных с едкими щелочами осуществлена бы-.ла незадолго до этого (1867 г.) одновременно и независимо друг от друга Кекуле, Вюрцем и Дюссором. Нафтольный щелочный плав был произведен Эллером в 1868 г. Другой интересный метод получения ализарина,
одновременно разработанный, с одной стороны, Грэбе и Либерманом, с другой—Перкином, запатентован, последним в Англии в 1869 г., состоит в том, что образующийся при хлорировании антрацена дихлоран-трацен при нагревании с серной кислотой переходит в дихлордисульфокислоту антрацена. Грэбе и Либерман дальнейшим нагреванием с серной к-той переводят это соединение в дисульфокислоту антрахинона, Перкин достигает того же эффекта окислением перекисью марганца. Метод этот одно время применялся технически, давая гл. обр. пользующийся большим спросом антрапур-пурин (1, 2, 7-триоксиантрахинон). Значительное улучшение получения сульфокислот антрахинона достигнуто было в 1873 г. Кохом, химиком фабрики бр. Гессерт, применившим для сульфирования высокопроцентную дымящую серную кислоту, т. н. олеум. Фабричная разработка контактного метода получения серной кислоты в первой своей стадии несомненно связана с потребностью ализариновой промышленности в олеуме. Открытие это чрезвычайно благотворно повлияло как на чистоту, так и на выходы А. Вскоре (1875 г.) тем же Кохом впервые в технику щелочного плавления введены автоклавы, применение которых понизило t° реакции плава с 200—280° до 170—180°, повысив выходы до 100—105 ч. сухого А. на исходный антрахинон (прежде 1/9 этого количества). Уже с 1871 г. ализариновым фабрикам было известно, что для получения А. нужно сплавлять не дисульфо-, а моносульфокислоту антрахинона, и что дисульфокислоты в этих условиях дают не А., а смесь триоксиантрахинонов. Опубликовано это было Перкином лишь в 1876 г. Реакция идет по ур-ию: C14H702(S03Na) + + 3NaOH + 0-> С14Н602(ONa)2 + Na2S03 + + Н20. В отсутствии кислорода получается тот же А., но реакция идет с выделением водорода: CJ4H702(0Na)-f NaOH =
=C14H602(0Na)2 + H2. Как это ни странно, но добавление к щелочному плаву селитры как источника кислорода было впервые применено лишь в 1874 г., на швейцарской ф-ке А. Биндшедлера и Буша. Кох (на ф-ке бр. Гессерт) с 1875 г. в качестве окислителя применял хлорноватокислый калий.
Современные методы выработки А. Исходным продуктом служит чистый 99—99,5%-ный антрахинон, подвергающийся сульфированию. Смотря по тому, желательно ли получить в главной массе моно- или дисульфокислоту антрахинона, сульфирование ведут либо при более низкой, либо при повышенной t° с меньшим или большим избытком ангидрида серной к-ты(20—40%-ный олеум). При сульфировании с целью получения 2-моносульфокислоты часть взятого в реакцию антрахинона (от 7з до у2) оставляют непросуль-фированной. Операцию ведут в закрытом железном котле, емкостью в 2 м3, с паровой рубашкой и мешй, с приспособлением для выдавливания массы сжатым воздухом.
Щелочные плавы ведутся в железных автоклавах, с мешй и масляной рубашкой, на 12 atm рабочего давления, емкостью до 10 м3 и более. Железо для ализариновых автоклавов должно быть по возможности свободно от фосфора. Такие автоклавы служат десятки лет. По окончании плава в котел накачивают воду, массу выдавливают в железные разварные баки и осаждают серной к-той. Высадившийся в виде желтых хлопьев А. фильтруют через деревянный фильтр-пресс и промывают до нейтральной реакции. Хорошая серебристая соль дает А. синеватого оттенка, не требующий дальнейшей очистки. Плавы для получения антрапурпурина и флаво-пурпурина с дисульфокислыми солями ведутся в общем аналогично плаву серебристой соли, но для получения А. хорошего качества и для увеличения выходов количество едкого натра повышают приблизительно вдвое. Растворение и осаждение плава ведется, как в случае А. синеватого оттенка. Как самый А., так и флаво- и антрапурпурин обладают чрезвычайно интересным свойством: при обработке известью в присутствии избытка едкой щелочи они количественно выпадают в виде кальциевых лаков, тогда как некрасящие примеси плава — монооксиантрахинон, изоантра- и антрафлавиновые кислоты — остаются в· этих условиях в растворе в виде натриевых солей. Это позволяет вести обработку плава с регенерацией едкой щелочи при одновременном получении чистого красителя плава.
Известкование флавопур-пуринового плава. 10 ж3 нагретого раствора щелочного плава флавопур-пурина, соответствующие приблизительно 300 килограмм А. обыкновенной желтой марки, обрабатываются 130 — 140 килограмм извести. Через % часа добавляют водой до 15 м3 и при 70° фильтруют через железный фильтр-пресс. Полученный после промывки горячей водой темносиний кальциевый лак флаво-пурп урина замешивается с водой и осаждается соляной к-той. Высадившийся фла-вопурпурин фильтруют, промывают. Выход сухого чистого флавопурпурина 220 килограмм. Свободный от флавопурпурина яркокрасный щелочной раствор (20° Вё) при охлаждении выделяет яркокрасный кристаллический антрафлавиновокислый натрий, соответственно 72 килограмма антрафлавиновой кислоты (смотрите). С целью регенерации едкого натрия остаточный щелочной раствор выпаривают сначала до 39° Вё, затем отстаиванием освобождают от выпавших солей (сульфат, сульфит), после чего окончательно концентрируют до 45°Вё. Такой окрашенный в красный цвет щелок содержит до 64 объёмных % NaOH и вполне пригоден для ализариновых плавов. Выход обратно полученного едкого натра—около 75% на исходное количество. Метод получения А. щелочным плавом непосредственно из антрахинона вследствие плохих выходов (распад молекулы до бензойной к-ты) не получил технич. применения, несмотря на то, что получаемый т. о. А. не содержит примесей триоксиантрахинонов и является поэтому продуктом чрезвычайно ценным.
Некоторые характерные свойства А. Как А., так и антра-и флавопурпурин возгоняются (значительные потери вследствие обугливания) в оранжево-красных иглах; они б. или м. хорошо растворяются в органич. растворителях, выделяясь при охлаждении в иголочках, но очень трудно растворимы в воде (растворимость А. в воде: 100 8 воды при 100° растворяют 0,0034 8, а при 250° — 3,16 г); в слабых щелочах они растворяются с характерной красно-синей окраской,— более синей для А., наименее синей для флавопурпурина; антрапурпурин занимает среднее место (колориметрическая проба). Соответственно с этим и окраски ализаринами по глиноземо-кальциевой протраве располагаются в таком же порядке, в смысле большей или меньшей синевы оттенка: А. дает красно-синеватые, антрапурпурин—наиболее красные, флавопурпурин — красно-желтоватые окраски. На железной протраве А. дает темнофиолетовые окраски, антра- и флавопурпурин—красноватофиолетовые. Характерные фиолетово-коричневые окраски получаются на хромовой протраве. О чрезвычайно характерном для А., антра- и флавопурпурина свойстве выпадать в виде кальциевых лаков в присутствии огромных избытков едкой щелочи говорилось выше. Точки плавления: А. 290° (диацетилпроизводное 184°), антрапурпурин 369° (триацетилпроизв. 224°) флавопурпурин 360° (триацетилпроизводное 195—196°). При сульфировании А. 20%-ным олеумом получается 3-сульфокислота А., натриевая соль которой СО ОН
ОН
1 4/Ao3Na
СО
имеет применение в крашении шерсти, давая на глиноземной протраве прочные ярко-красные оттенки. По хромовой протраве получаются прочные красно-коричневые окраски. Сульфокислота эта не образует 1,2, 3-триоксиантрахинона (антра-галлола) при сплавлении со щелочами, регенерируя ализарин. Так называемый А. оранжевый, 3-н итроализарин СО ОН
он
Ilii
/no2
со
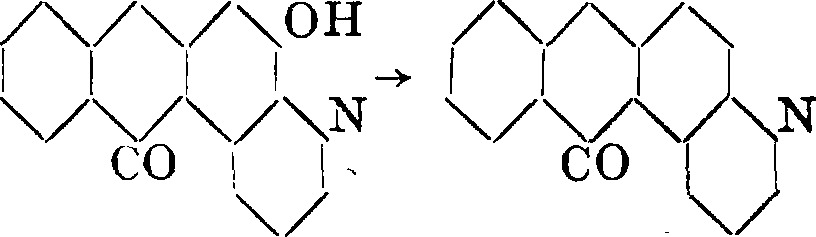
образуется при нитровании А. в крепкой серной кислоте, в присутствии борной к-ты. Соединение это на глиноземной и хромовой протраве имеет применение в печати. При восстановлении образуется 3-амино-ализарин, переходящий при конденсации с глицерином, в присутствии крепкой серной кислоты и окислителей, в А.-х и-н о л и и, т. н. ализариновый синий, который в виде бисульфитного соединения имеет значит, применение в печати. При нагревании с цинковой пылью А.-хинолин переходит в β-a нтрахинолин
СО ОН со
Интересно, что ализариновый синий окрашивает бумажное волокно в синий цвет также из куба, являясь, таким образом первым искусственным кубовым красителем. А.- бордо (1,2,5, 8-т етраокси-антрахинон) получается с хорошими выходами осторожным окислением А. высокопроцентным олеумом. 10 ч. А. обрабатываются при 40—50°, в течение 3—5 дней, 120 ч. 80%-ного олеума. Сначала выпадают кристаллы эфира серной к-ты, омыляю-щегося при нагревании с водными щелочами или к-тами. То же самое тетраокси-производное образуется и при окислении хинизарина; отсюда вытекают и формулы строения этих красителей:
СО ОН он со он он он
VVV он со
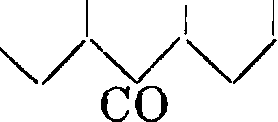
он со
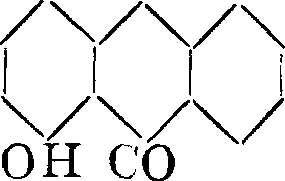
почему это соединение и носит татке название хинализарина. По глиноземной протраве А. - бордо дает сытый бордо, по хрому—сине-фиолетовый оттенок. Он хорошо применим и для крашения животного волокна. Пурпурин технически получается окислением А. перекисью марганца в крепкой серной кислоте или олеуме в виде длинных оранжевых игол, содержащих 1 молекулу воды и плавящихся при 256°. Дает по глиноземной протраве живые красные оттенки, не отличающиеся особой прочностью; поэтому пурпурин непосредственно как краситель не применяется. Очень ценно производное пурпурина—так называемым А.-с ине-черный для крашения шерсти, получаемый конденсацией пурпурина с анилином, с последующим сульфированием продукта.
Современное значение А.: более или менее серьезные конкуренты А. начинают появляться на рынке уже с середины 80-х гг. Появление т. н. пара-красного (п-нитранилин — β -нафтол) оказало большое влияние в смысле уменьшения спроса на А. Правда, пара-красный не отличается светопрочностыо,но светопрочность требуется не во всех случаях; гораздо более серьезным конкурентом А. является т. н. нафтол АС, в комбинации с новыми основаниями, как нитроаминоанизол и тому подобное. Такие окраски, отличаясь хорошей прочностью и прекрасною способностью к выправлению, в общем все же во многом уступают А., дающему окраски исключительной прочности по отношению к действию света и воздуха. К тому же А. принадлежит к числу дешевых красителей, тогда как новые красители нафтол АС и их компоненты расцениваются на рынке раза в три дороже и, по сложности химического состава, едва ли когда-либо станут дешевым продуктом. Крупным недостатком А. является бесспорно его сложный и дорогой способ окраски, правда, в настоящее время уже сильно упрощенный, но, к сожалению, в ущерб прочности окрасок. Если удастся разработать скорый и дешевый метод прочного ализаринового крашения, то потребление А. сейчас же опять повысится.
Следует иметь в виду, что А. дает не только так называемый адри анопольский красный, но, путем комбинирования как самого ализаринового красителя, так и различных протрав, любой прочный оттенок, от желтого до темнокоричневого и сине-черного. Следует привести отзыв известного специалиста, проф. Цюрихского политехникума Фирц- Давида, указывающего в своем руководстве на значение А., наряду с индиго являющегося самым значительным продуктом химии красок. Уже через год после открытия Грэбе и Либерма-на, в Англии на ф-ке Перкин и с-вья началась техническая выработка синтетического А., затем в том же году—на 6 германских ф-ках.Число фабрик, вырабатывающих А., быстро возрастает до 18 (12 германск., 2 швейц., 2 англ., 1 франц. и 1 австр.), сократившись к 90-м гг. до 12 (8 в Германии, 1 в Швейцарии, 2 в Англии и 1 в России). В начале текущего столетия число ализариновых фабрик, вследствие заключения ализариновой конвенции, сократилось до 5 (Баден, анил. и сод. ф-ка, Мейстер Люциус и Брюнинг, Ф. Байер и К°, Р. Ведекинд и К° в Германии и Британская ализариновая ф-ка). Остальные ализариновые заводы принуждены были прекратить выработку. К 1910—1911 гг. снова проявляется тенденция к пуску новых ализариновых фабрик— в Германии Грисгейм-Электрон и Магде-бургская ф-ка, в Австрии — в Ауссиге. В России А. стал вырабатываться еще в начале 80-х гг., в Лобановке, под Москвой (Биндер и Беме). Завод этот вскоре перешел к фирме Л. Рабенек, в Щелкове, под Москвой. А. прекрасного качества, в количестве около 100 тонн в год (100%-ный А.), вырабатывался этой фирмой в течение 15 лет, вплоть до 1899 г. В 1870 г., ко времени изобретения синтетического метода получения А., выработка его из краппа выражалась приблизительно в цифре 750 тонн (100%-ный А.).
Выработка синтетического А.
(100%-ный А).
в 1872 г. достигала.. 50 m
» 1 873 » » 100»
» 1 874 » » 130»
» 1 888 » » .. 1 820 »
» 1896 » » .. 3 000 »
с 1900 » по 1914 г. достигала до. 2 800 »
Цена на А. за 1 килограмм (100%-го).
в 1871 г. из краппа.80 герм. мар.
» 1873 » синтетический.140 » »
» 1888 » ». 8 » »
» 1 896 » ». 5,50 » »
с 1 900 » по 1 914 г. спит. .4,30 — 5,50 » »
Довоен. выработка А. составляла в круглых цифрах в Германии 2 000 тонн 100%-го и в Англии 1 000 ж 100%-го, всего 3 000 ж. Выработка ализаринового завода Л. Рабенек в Москве равнялась ~ 100 тонн годовых (100%-го). Годовая довоенная потребность России в А. равнялась 410 ж (100%-го), около х/7 мирового потребления. В настоящее время А. вырабатывается, кроме Германии, в Англии, Франции, Италии, Чехо-Слова-кии и в С.-А. С. Ш. Каких-либо точных данных о сравнительных выработках в этих странах А. не имеется. У нас пуск ализари нового завода Анилтреста (на 200 ж 100%-го о предполагается к весне 1928 г.
Лит.: Г е о р г и е в и ч Г. и Грани ужен Е., Химия красящих веществ, М., 1916; Шапошников В. Г., Общ. технология волоки, и красящих веществ, BTC, М.—Киев, 1926; Энцикл.сл. Брокгауз-Ефрон, т. 2, Лейпциг—СПБ., 1911; Auerbach, G., Anthracen u. seine Derivate, Braunschweig, 1880; Schultz G., Die Chemie des Steinkohlenteers, Braunschweig, 1926; Gnehm R., Die Anthracen-farbstoffe, Braunschweig, 1897; Meyer V. u. Jacobson P., Lehrbuch d. organiscli. Chemie, B. 2, T. 1, Lpz., 1903; Ullmann· F., Enzyklopadie d. techn. Chemie, В. 1, Wien, 1914; F i e г z-D a v i d H„ Kunstliche organ. Farbstoffe, B., 1926; Wiirtz A., Dictionnaire d. Chimie pure et appliqu6e, v. 1, 2 et Supplement 1, P., 1868—1880. M. Ильинский.