 > Техника, страница 12 > Аммиак
> Техника, страница 12 > Аммиак
Аммиак
Аммиак, NH3 мол. в 17,03. При коми. t° бесцветный газ, раздражающий слизистые оболочки. А. легко сгущается в жидкость, которая кипит при —33°,4 и закристал-лизовывается при —77°,3. Чистый сухой А. является слабой к-той, что ясно из возможности замещения в нем водорода натрием и образования амида натрия NH2Na
| Упруго | с т ь | пара | и; и к к | ОГО | а м м и | а к ί | |
| — 77’,9 | 4 | 4,9 | ИМ | + | 10’ | 6,085 | aim |
| — 70° | 0, | 171 | αί7η | + | 20’ | 8,459 | » |
| — 50" | 0, | 403 | » | -L | 30’ | 11,512 | » |
| — 30’ | 1, | 180 | t> | + | 40’. | 15,339 | » |
| — 20° | 1, | 877 | » | + | 50’ | 20,059 | » |
| — 10’ | 2, | 870 | + | 60° | 25,797 | » | |
| 0’ | 4, | 238 | » | + | 70’ | 32,687 | » |
при нагревании Na в струе А. Однако А. чрезвычайно легко присоединяет воду и образует щелочь NH4OH, едкий аммоний; раствор едкого аммония в воде называется нашатырным ом. Наличие А., улетучивающегося из едкого аммония благодаря разложению NH*OH^NH, + НОН, открывается по посинению лакмусовой бумажки. А. легко присоединяется к к-там, образуя соли NH4. Hanp.NH3+ HC1=NH4C1, что заметно, если пары А. (из нашатырного а) и пары НС1 встречаются в воздухе: тотчас образуется белое облачко нашатыря ИНДЛ. А. применяется обычно в виде нашатырного а (JD=0,91, ок. 25% NH3) и так называемым «ледяного нашатырного а» (D=0,882, с 35% NH,).
Крепость нашатырного а проще всего определять по его плотности, величины которой приведены в следующей таблице:
Плотность растворов аммиака (Л у п г е и Верни к).
| D при 15’ | NH, (в % |
| 1 ,000 | 0,00 |
| 0,998 | 0,4 5 |
| 0,996 | 0.91 |
| 0,994 | 1, 37 |
| 0,992 | 1, 84 |
| 0,990 | 2,31 |
| 0,9 86 | 3,30 |
| 0,982 | 4,30 |
| 0,980 | 4, 80 |
| 0,974 | Η. 30 |
| 0,970 | 7,31 |
| 0,96 8 | 7.82 |
| 0,966 | • 8,33 |
| 0,964 | 8,8 « |
| 0,962 | 9,35 |
| 0,960 | 9,91 |
| 0.958 | 10,47 |
| 0,954 | 11,60 |
| 0,9 52 | 12,17 |
| 0,950 | 12,74 |
| 0,948 | 13,31 |
| 0,946 | 13,88 |
| 0,944 | 14,46 |
| 0,942 | 15.04 |
| 0,940 | 15,63 |
| 0,938 | 16,22 |
| 0,9 36 | •16,82 |
| 0,934 | 17,42 |
| 0,9 32 | 18,03 |
| 0,930 | 18,64 |
| 0,926 | 19,87 |
| 0,922 | 21,12 |
| 0,920 | 21,75 |
| 0,914 | 23,68 |
| 0,910 | 24,99 |
| 0.908 | 25,6 5 |
| 0,906 | 26,31 |
| 0,904 | 26.98 |
| 0,902 | 27,65 |
| 0,900 | 28,33 |
| 0,898 | 29,01 |
| 0,894 | 30,37 |
| 0,892 | 31,05 |
| 0,890 | d 1 j j а |
| 0,888 | 32.50 |
| 0,8 86 | 33,25 |
| 0.884 | 34.10 |
| 0,882 | 34,95 |
| В 1л. Nil. при 15’ (в г) | Поправка па -- 1 |
| 0,0 | 0,00018 |
| 4,5 | 0.00018 |
| 9,1 | 0,00019 |
| 1 3, G | 0,00019 |
| 18,2 | 0.00020 |
| 22,9 | 0,0002(1 |
| 32,5 | 0,00021 |
| k 2 2 | 0,00020 |
| 4 7,0 | 0,0002 3 |
| 6 1.3 | 0,00024 |
| 7 0.9 | U,00025 |
| i 5, t | 0,00 02.6 |
| 80,5 | 0,00026 |
| 85,2 | 0,00027 |
| 89,9 | 0,00028 |
| 95, 1 | 0,00029 |
| 100,3 | 0,00030 |
| 110.7 | 0,00032 |
| 115,9 | 0,00033 |
| 121,0 | 0,00034 |
| 126,2 | 0.00035 |
| 131.3 | 0.00036 |
| 136,5 | 0,00037 |
| 1 4 1 ,7 | 0,00038 |
| 146,9 | 0,00039 |
| 152.1 | 0,00040 |
| 157,4 | 0,00041 |
| 162,7 | 0,00041 |
| 168,1 | 0,00042 |
| 173,4 | 0,00042 |
| 184,2 | 0,00044 |
| 194,7 | 0,00046 |
| 200,1 | 0,00047 |
| 216,3 | 0,00050 |
| 227,4 | 0,00052 |
| 2 32,9 | 6,00053 |
| 238,3 | 0,00054 |
| 24 3,9 | 0,00051 |
| 249.4 | 0,00056 |
| 255,0 · | 0,00057 |
| 260,5 | 0,00058 |
| 271,5 | 0,00060 |
| 27 7.0 | 0,00060 |
| 282,6 | 0,00061 |
| 288,6 | 0,00062 |
| 294,6 | 0.00063 |
| 301,4 | 0,00064 |
| 308,3 | 0.00065 |
i
Упругость пара водных растворов А. слагается из парциальных упругостей А. и воды, приведенных в таблице:
| У п | р у Г о с | т ь | п | ара вод | Η | ы х pa | с т в 0 | а о
ft | |
| t° | % NH, | р NH, | V Н20 | i 0 | % NH, | Р NH, | р Н.О1 | ||
| по весу | (в .(Of) | (в .мм) | по весу | (в миллиметров) | (в ΛΟΙ) | ||||
| 0° | 9,15 | 2 4 | 8 | 5,3 | 19 · | 9 | 10,15 | 80,6 | 15,1 j |
| 14,73 | 51 | ,3 | 4,1 | 16,64 | 166,1 | 12,9 | |||
| 22,90 | 116 | 6 | 2,8 | 23,37 | 302,4 | 10,3 | |||
| 10’ | 12,32 | 64 | ,2 | 7,6 | 50 | 11,57 | 341,7 | 80,6 | |
| 15,88 | 95 | ,1 | 7,0 | i | 14,94 | 87,1 | 75,2 | ||
| 21,83 | 169 | ,8 | 5,5 | 1 | 1
1 | ||||
Понятно, что упругость пара А. как вещества, кипящего при t°, значительно низшей, чем t°Kun. воды, > парциальной упругости паров воды над нашатырным ом. Растворимость NH3 в воде очень велика.
Растворимость аммиака η воде.
I
ί
I Коэфф. абсорбции. Число объём, газа (0 I и 7 60 aot), поглощ. i при парц. давл.
I 760 миллиметров 1 объёмом i растворителя
I Число а аммиака, i поглощаемого 100 г воды, если общее ί давление | ; (газ+вода) 760 миллиметров j
| 0° | 1 176 | 89,5 |
| 8’ | 9 47 | 72,0 |
| 12 | 8 5 7 | 65,1 |
| 1 3 | 8 87 | 0 3,6 |
| 1 6 · | 775 | 5 8,7 |
| 20 ’ | 702 | 53,1 |
| 24’ | 639 | 48,2 |
Однако при абсорбции NH3 водой мы имеем дело не только с процессом растворения, но и с образованием гидроокиси А.; эта гидроокись содержится в нашатырном е и легко диссоциирует но уравнению NH4OH ^ NH3 + Н20. В виду этого NH3гораздо слабее как щелочь, чем NaOH или КОН, хотя образует весьма прочные аммонийные соли, например NH3+HC1=NH4C1; при этом А. непосредственно соединяется с к-тами (об аммонийных солях см. Аммония соединения). Нашатырный широко применяется как щелочь в лабораториях, в нек-рых производствах и для получения соды по Сольвею. Соли А. слабых к-т сильно гидролизованы и пахнут поэтому А. (например углекислый и борнокислый А.). Кремнекислый А. уже непосредственно после получения обменным разложением выделяет кремневый студень и А. Способность А. образовывать комплексы чрезвычайно велика. К ним относятся очень прочные [Со(NH3)e] Cl3, [Cr(NH3)e] Clg, [Pt (NH,),] Cl4; очень прочными являются аммиачные комплексы меди и никеля в водном растворе; их легко узнать по чрезвычайно интенсивному окрашиванию. При действии на галоидозамещенные углеводороды А. дает последовательно первичные, вторичные, третичные амины и замещенные производные аммония. Из всех хим. соединений А. в чистом виде по целому ряду свойств наиболее сходен с водой. Он имеет очень высокую t°Kpum. (132°,53) и очень высокое критическое давление (115 atm), то есть в этом отношении столь же отклоняется от обычных норм, как и вода. Теплоемкость жидкого А. даже выше, чем у воды, а именно:
С0=1,152 и С — 1,172. Все это указывает на очень высокую величину ассоциации (смотрите) А. Способность последнего образовывать комплексы с неорганич. ионами и высокая величина ассоциации обусловливают растворимость многих солей в нем. Это обстоятельство дало материал для ряда интересных научных исследований, особенно в области электропроводности; в дальнейшем это свойство А. может быть широко использовано в технике.
А. выделяется при вулканических извержениях, содержится в парах фумарол и горячих источников Тосканы. При анаэробном гниении всегда образуется А., равно как и при медленном разложении многих органических веществ. Добыча А. из человеческих отбросов распространена во Франции, но выгодна лишь тогда, когда завод может получить от города в год от 50 000 до 100 000 м3 фекалий, при населении в 50 000 жителей. Нормально должно было бы быть
7—8 килограмм А. в 1 Λΐ3; на деле, благодаря разбавлению промывочными водами и всасыванию воды извне, из 1 jh3 фекалий получается
3,6—4,4 килограмма А. или 14—17 килограмм сернокислого аммония и 10—15 килограмм фосфорного пудрета. Мочевина, составляющая свыше 90% азотистых веществ мочи, под влиянием бактерий переходит в углекислый аммоний, который легко переработать на А., но из мочевой и гиппуровой к-т, креатина и ксантина можно получить А. лишь кипячением с известью. По старому способу применяли для перегонки лишь воду из открытых отстойников; твердые остатки сушили, прессовали и продавали под названием пудрета. Теперь обработку как жидкости, так и твердых остатков ведут в закрытых сосудах, где сначала все смешивают, отделяют твердые куски, например битое стекло, жестянки и тому подобное.; затем массу переводят через подогреватель в перегонные аппараты, где отгоняются А. и сероводород, после чего добавляют извести и заканчивают перегонку. А. улавливают в коксовой башне, орошаемой серной к-той. Твердый остаток высушивается и превращается в пудрет с 7—8% фосфорной кислоты и 1—2% связ. азота. В 1905 г. во Франции получили из человеческих отбросов до 9 000 тонн сернокислого аммония, в 1913 г.—свыше 12 000 тонн Кроме того, во Франции пользуются животными твердыми остатками, например обрезками и грязью боен, рынков, костями и тому подобное. Однако часто А. не выделяют, но ограничиваются очисткой отбросов от механич. примесей, прокаливают их в автоклаве и высушивают в пустоте (прибавив предварительно серной к-ты, чтобы связать А.). Кости, рог, отбросы, в роде волос, кожи и тому подобное., содержат много азота (в роге до 16%), и при сухой перегонке этого материала для получения животного угля можно получить до 250—300 килограмм сернокислого аммония на м3 исход, материала.
Главным источником А. долгое время являлся каменный уголь, содержащий до 1,94% азота. При сухой перегонке каменного угля 10—25% азота переходит в А., причем наибольшее количество А. получается, если перегонка ведется при 900°.
Высшие и низшие t° дают худший результат. Часть азота выделяется в свободном виде, а часть—в виде циана. В виду наличия в газах сухой перегонки каменного угля угольного ангидрида, сероводорода, хлоро-водорода и роданистого водорода, образуются соответственные соли аммония, причем соотношения их по весу крайне разнообразны, но больше всего получается углекислого аммония и нашатыря. Поглощение А. на газовых заводах производится или в специальных промывных аппаратах в виде барабана, наполовину наполненного водой, с мешалками, или в специальных промывных колонках. При больших установках весь А. задерживается в промывате-лях или в колонных аппаратах. При сухой перегонке каменного угля на кокс поглощение ведут серной кислотой, что уменьшает осветительную способность газа, но зато из коксовых печей получают немного больший выход А., чем на газовых заводах. А. возможно улавливать и из газогенераторов, причем, если t° держать достаточно низкой и впускать пар, перегретый до 250—380°, то можно получить до 4 килограмма А. на т. Если во время сухой перегонки каменного угля впускать водяной пар, подогретый до 150° и смешанный с воздухом, то при 1,5 тонн воды на 1 тонна угля выход А. соответствует 60—70% азота в угле. Т. о. в Англии перерабатывают около 1 000 000 тонн в год, причем на 1 m угля получают 40 килограмм сернокислого аммония. При переработке торфа по способу Вольперека влажный воздух пропускается при 450° над свежедобы-тым торфом; получается 5% сернокислого аммония, до 1,5% уксусной кислоты и 3—8% парафина (на 100 m сухого торфа 5 тонн сульфата). Если перед перегонкой к углю прибавить извести, то выходА повышаются на 20—40%. При перегонке барды (мелассы), ее сгущают до 70° В6 и прибавляют 0,5—1 % прокаленной глины, а затем подвергают сухой перегонке, причем, кроме А., получают заметное количество метиламина. Газовая вода, собранная в холодильниках при перегонке каменного угля, содержит в 1 л около 16—16,5 г А. и около 3 г серы, причем на 100 г А. приходится:
| ОКОЛО | 7,80 | г | соединенного | с | H.S |
| » | 78,80 | » | » | » | н,со, |
| 1> | 12,50 | » | У> | НС1 | |
| » | 0.70 | » | » | HCNS | |
| » | 0,24 | » | » | » | HsSO* |
| » | 0,67 | » | » | » | HjSjOi |
| » | 0,12 | » | » | HCN. |
Из них А., соединенный с H2S, HCNS, Н2С03 в виде солей, разлагаемых водой при кипячении, называется «летучим»; остальной А. в виде соединений, разлагаемых только известковым молоком,— «нелетучим». Количество А. определяется отгонкою в титрованную серную к-ту. Аммиачная вода коксовальных з-дов содержит, в силу слабого охлаждения и большого содержания влаги в угле, 8—15 г А. в л; на газовых з-дах получается газовая вода, содержащая 30—50 г. А. на л, т. к. уголь предварительно подсушивается и выделяет поэтому меньше влаги, а газы сильнее охлаждаются. Обычно газовую воду собирают в подземные резервуары и оттуда направляют ее в колонные аппараты для отгонки А. В середине колонны к воде добавляют известковое молоко; иногда колонну с известковым молоком ставят рядом с водной колонной. При перегонке в течение 24 ч. 30 JH8 газовой воды с содержанием
2,5—3% NH3 в отходящих водах остается не больше 0,005% NHS. Выделенный NH3поглощают серной кислотой или очищают при помощи Са(ОН)2 от примеси С02, затем пропускают через свежепрокален-ный древесный уголь, через NaOH и, наконец, пускают для растворения в воду. Примерно 10 ж8 газовой воды дают 700 килограмм нашатырного а с 25% NH3. А. возможно добывать из цианамида кальция CaCN2, получающегося путем присоединения азота к карбиду кальция (смотрите ниже Получение синтетического А.). Существует несколько способов для перевода азота цианамида в А. По способу Франка и Каро нужно вести нагревание с водяным паром при повышенном давлении: CaCN2-f -f 3H20=CaC03+2NH2. Эту реакцию проводят в больших стальных автоклавах (6,4 метров выс., 1,8 л в диам., 20 atm давл.). Цианамид разлагается либо раствором от прежней реакции, либо водой (5,5 л8), медленно, для удаления ацетилена; затем прибавляют немного соды и извести, автоклавы запирают и нагревают до 133—143°, то есть до давления в 3—4 atm водяного пара, но выделяющийся А. доводит давление до 15 atm. Один автоклав в 24 ч. дает 2,3 тонны А.; каждая операция с последовательным подогревом требует около 3—3,5 ч.; выход—ок. 96—98% на содержащийся в цианамиде азот. (Чертежи завода для переработки 75 000 тонн цианамида кальция в 350 дней приведены у Waeser, Die Luftstickstoif-Industrie.) Цианамид из запасных хранилищ по горизонтальному шнеку передается в ряд силосов, из них— в ряд небольших запасных резервуаров и через автоматич. весы — в 40 автоклавов, причем один шнек снабжает 5 автоклавов. Вода и щелочь подаются из верхних баков. В автоклавном помещении устраивают очень сильную вентиляцию для удаления цианамидной пыли и ацетилена. Выделяющийся из автоклавов А. охлаждается в 8 холодильниках и спускается в находящиеся под автоклавами испарители; там остаются вода и примеси, а газ снова направляется в конденсаторы и из них—в охладители, где и образует крепкий нашатырный. Его перекачивают в запасные резервуары. Берлин-Ангальтское машиностроительное общество (BAMAG) строит специальные аппараты для очистки А. Сырой А. направляется в испаритель, причем вода вытекает из последнего через нижнее отверстие. Смесь водяных и аммиачных паров проходит через колонный конденсатор и такой же запасный холодильник, откуда сгущенная вода возвращается в испаритель, а газы проходят через очиститель с едким натром. Из него они поступают в башню, для очистки от увлеченных частиц щелочи, и оттуда, через три колонны с древесным углем, в колонку для поглощения NH3 водой. В случае дешевизны бисульфата натрия (во время войны Германия име ла до 70 000 тонн в месяц в виде отброса) возможно получить более чистый А. из цианамида по реакции:
2CaCNi+2HsO=Ca(OH),+ Ca(HCNa),;
Са( О Н)2 + Са( Н CN2)2+2N aHS04=2 CaS 04 -+ +2H20 + 2NaHCN2;
NaHCN,+3HiO=NaHCO,+2NHs;
2NaHC03=Na2C03+H20+C02;
2NaHCN„+2Ca(0 H)2+4H20=2NaOH+ +4N H3-f 2CaC03.
Бисульфат NaHSO, доставляется как отброс от добывания азотной кислоты, причем описанный способ дает очень чистый NH3.
Чтобы дать понятие о соврем, производстве А. из цианамида, достаточно указать, что на з-де в Пистерице в 24 ч. добывается 330 тонн карбида и 430 тонн цианамида. Восемь карбидных печей поглощают 60 000 kWh, 32 вагона извести, 22 вагона кокса. Азотирование производится в 720 печах, азот подается 6 компрессорами Линде, добывающими из 210 000 м3 воздуха 150 000 ж8 чистого азота. Печи дают в среднем 430 тонн цианамида с 18—20% азота. В 24 ч. потребляется 100 000 ж8 воды. 20 автоклавов потребляют 210 тонн цианамида и дают 50 тонн КН3, к-рый в 10 поглотителях дает 200 тонн сульфата или 200 тонн 25%-ного А. Нитридный способ Сер-пека, основанный на поглощении алюминием азота в момент восстановления:
A1203+3C+2N=2A1N + 3C0, и разложении A1N водой:
2A1N + 6HS0^2NH3+2A1(0H)3,
не получил еще широкого технич. применения, хотя его большим преимуществом является дешевое получение чистой окиси алюминия для алюминиевого производства. Громадные количества А. добываются в наст, время синтетически из азота и водорода по способам Габера—Боша, Казале и Клода (смотрите Азот и ниже Получение синт. А.). Чистый А., безводный, потребляется в больших количествах для холодильных машин и для пересылки вместо нашатырного а для удешевления транспорта. А. продается в стальных цилиндрах и должен содержать не больше 0,1% примесей.
Лит.: Ост Г., Химич. технология, Л., 1924·—27; Fr. Ullmann’s Enzyklop&die d. techn. Chemie, B.— Wien, 1914 u. ff.; В e 11 z e r F., La chimie indu-strielle moderne, t. 1—2, P., 1909—1 1; Waeser В r. Die Luftstickstoff-Industrie, Lpz., 1922.
Получение синтетического А. А. получается непосредственно из элементов N и Н, при высокой t° и высоком давлении, при участии разных катализаторов. Реакция соединения N2 + ЗН2 ^ 3NH3 + 22 000 cal является процессом экзотермическим и при нормальном давлении и обыкновенной t* должна была бы протекать почти нацело в смысле образования NH3. Но чрезвычайная инертность азота при нормальной t° обусловливает чрезвычайно малую скорость этой реакции, для ускорения которой необходимо значительно повысить t°. При высокой t° (при t° красного каления) реакция, правда, идет очень быстро, но равновесие N2 -f ЗН2 ^ 2NHS устанавливается при совершенно ничтожном содержании NH3 в газовой смеси. Чтобы ускорить процесс и заставить его пройти при более низкой t°, соответствующей более значительному содержанию NHS в смеси, можно было бы воспользоваться действием какого-нибудь катализатора, но ни один из известных катализаторов не оказывает сколько-нибудь заметного действия на реакцию синтеза NH, из элементов при сравнительно невысокой t°. Поэтому естественно было подумать о том, чтобы высокую t° в процессе синтеза А. при участии катализаторов понизить, а высокой концентрации NH3 добиться путей увеличения давления. Приводимая ниже таблица, опубликованная Ларсоном (1924 год), работающим в Правительственной лаборатории по исследованию азота в С.-А. С. Ш., прекрасно иллюстрирует зависимость процентного содержания NH3 в указанном равновесии от разных условий t° и давления.
Процентное содержание NH, в равновесии Na + 3H, ^ 2NH3.
Давление в atm
| 30 | 100 | 300 | 600 | 1000 | |
| 20(Г. | 6 7,65 | 81,65 | 89,94 | 95,37 | 98,29 |
| 300°. | 30,25 | 52,04 | 70,96 | 84,21 | 92,55 |
| 400’. | 10,75 | 25,12 | 47,00 | 65,20 | 79,82 |
| 5005. | 3,49 | 10,61 | 26,4 4 | 42,15 | 57,47 |
| 600°. | 1,39 | 4,52 | 13,77 | 23,10 | 31,43 |
| 700°. | 0,08 | 2,18 | 7,28 | 12,60 | 12,87 |
Теоретически идея применения высоких давлений для синтеза NH3 из элементов была подготовлена уже давно целым рядом исследователей, среди которых необходимо особенно выделить немецк. химика Йоста, который в лаборатории В. Нернста установил как теоретически, так и экспериментально решающее значение повышения давления на реакцию синтеза А. из элементов. Однако работы Йоста и самого Нернста не обратили на себя в свое время должного внимания, и только упорные исследования в том же направлении выдающегося немецкого химика Габера и его учеников и вместе с тем живой интерес и поддержка, проявленные к реакции синтеза NH3 «Баденским анилиновым и содовым производством» (BASF), дали возможность Габеру совместно с Бошем (инж. BASF) довести дело до конца и реализовать в технич. масштабе величайшее достижение химич. технологии всех времен — рентабельный синтез связанного азота в виде NH3 из элементов, то есть фиксацию свободного азота атмосферы. На пути к осуществлению этого синтеза в заводском масштабе встали исключительные трудности технич. характера, в преодолении которых главная роль принадлежит инж. Бошу, Митташу и др. Эти трудности заключались, прежде всего, в необычной для хим. заводских установок необходимости конструировать аппаратуру для проведения химич. процесса при столь значительном давлении (до 200 atm и более); а между тем именно такое давление требовалось для получения достаточного выхода при реакции синтеза NH3 из элементов. Опасность ов и разрушения аппаратуры усугублялась еще тем, что реакция должна была проводиться все vice
при достаточно высокой t° (550°). При этом выгодная сторона реакции, ее экзоте рмичность, в данном отношении также создавала трудности в смысле необходимости тщательного регулирования термической стороны процесса. Значительные трудности возникли также вследствие того, что уже небольшая примесь кислорода к содержащей водород газовой смеси, в условиях столь высокого давления и t°, обусловливала сильные ы гремучего газа, вследствие чего необходимо было установить автоматический контроль над пускаемой в реакцию смесью газов и тщательно оберегать ее от проникновения в нее кислорода. Реагирующая смесь газов должна была предварительно очищаться не только от присутствия кислорода, но и от присутствия хотя бы следов целого ряда других веществ — сернистых, овистых соединений и в особенности от присутствия окиси углерода, которая к тому же, как увидим далее, входя в исходный продукт для получения водорода из водяного и коксогенераторного газов, почти неизбежно содержалась в добываемом водороде. Все эти примеси, даже в ничтожных количествах, «отравляли» контактную массу, хранимую в сокровеннейшей тайне. Весьма нелегкую задачу представило подыскание состава этой контактной массы, причем пришлось исследовать огромное число различных катализаторов и их комбинаций, прежде чем был установлен дешевый достаточно активный катализатор и изучены условия его действия и «отравления» разными ядами.
Но особенные, совершенно исключительные трудности при осуществлении синтеза NH3 из элементов возникли в связи с тем, что водород при столь высоких давлениях и температурах, во-первых, диффундировал через лучшие сорта стали, из которых изготовлялись печи высокого давления, необходимые для процесса, и, во-вторых, взаимодействовал с углеродом стали, обез-угливал сталь и делал ее хрупкой и чрезвычайно подверженной разрывам. Только упорство и исключительное искусство Боша и его сотрудников позволили им подъ-искать соответствующий металлич. материал для изготовления вых печей («облагороженная» примесью 18% W и 3% Сг сталь с незначительным содержанием С, или даже свободная от последнего, и другие сорта стали). Аппаратура для проведения синтеза NH3 была сконструирована Бошем и его сотрудниками так. обр., чтобы давление газов внутри контактного, сосуда уравновешивалось снаружи равным ему противоположным давлением охлаждаемого газа, нагнетаемого в толстостенный защитный металлический футляр, в котором помещался собственно реакционный контактный трубчатый сосуд. В качестве исходных материалов метод Габер а—Б о ш а применяет смесь N2+3H2 в стехиометрическом количественном отношении (1 : Зпо объёму), получаемую из водяного газа (смотрите) и генераторного газа. Образовавшийся в количестве δ% NH3 поглощается водой под давлением в 200 atm. Непрореагировавшая смесь азота и водорода циркуляционным насосом нагнетается обратно в контактный сосуд с добавлением нового количества смеси исходных газов взамен образовавшегося и поглощенного NHS. На фигуре 1 изображена схема установки метода Габера— Боша в первоначальной конструкции, при
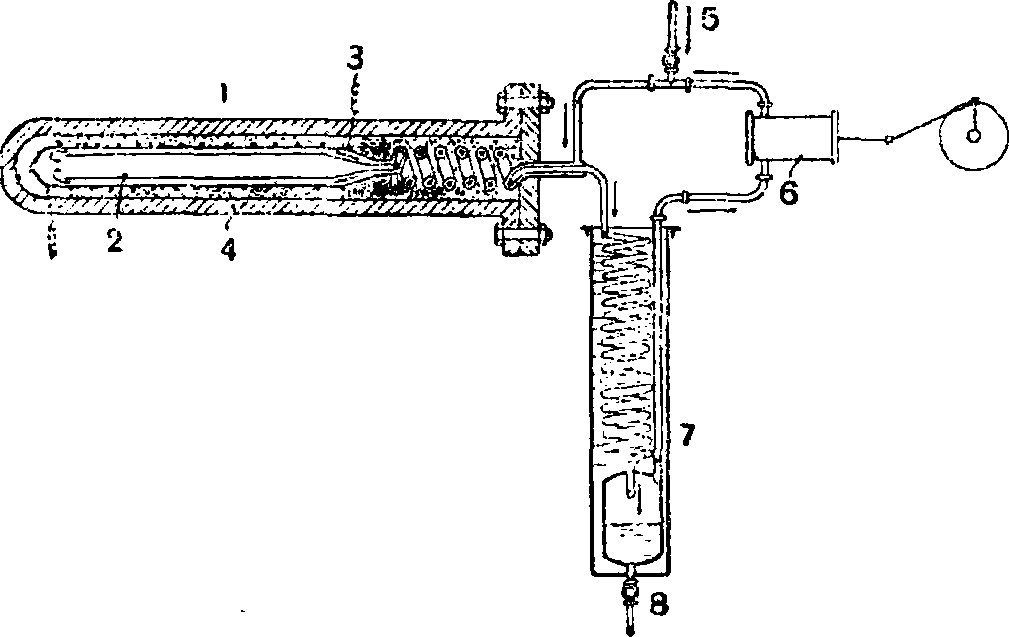
Фигура 1. Схема установки Габера—Боша: 1—лечь, г—контактная камера, г—электрический нагреватель, 4 — защитный толстостенный железный кожух, 5—приток нового газа (N+ЗН), в—циркуляционный насос, 7 — холодильник. S—спуск конденсированного аммиака. которой NH3 не поглощался водой, а сгущался в жидкий NH3 путем охлаждения. Этот путь изъятия готового NH3 практикуется и ныне в несколько видоизмененном методе Казале, по к-рому синтез осуществляется при давлении 750 atm. При таком давлении достаточно охлаждения до 20—25°, чтобы NH3 сгустился в жидкость. Описанный метод Габера—Боша реализован в Германии в двух крупнейших в мире предприятиях Берлинского азотного синдиката, а именно: в Оппау—BASF, с производством в 1924 году до 100 000 тонн связанного азота, и в Мерзебурге (Leunawerk «Аммиачных заводов»), с производством в 25 000 тонн связанного азота. Оба предприятия вырабатывают не только NH3, но и сернокислый аммоний, мочевину и другие азотные удобрительные продукты.
Кроме метода Габера — Боша, для синтеза NH3 из элементов заявлено еще несколько патентов, представляющих собой, в сущности, видоизменения того же самого метода. 1) Метод Казале характеризуется применением давления до 750 atm и меньшими аппаратурными «единицами» установок, большим выходом NH3 и выделением его в жидком виде путем охлаждения до 20°. В качестве исходного продукта применяется электролитический водород. По этому методу уже работают и, кроме того, еще организуются небольшие установки во Франции, Италии, Бельгии, Швейцарии, Испании, С.-А. С. Штатах и Японии, — всего с производительной способностью в 521,5 тонн связанного азота в день (главным образом во Франции и Японии). 2) Метод Клода применяет давление в 900 atm, причем продукт получается с содержанием 25% NIL. При методе Клода исходный материал, "коксогенераторный газ, последовательно попадает в различные камеры высокого давления, в которых сжимается; пять таких камер образуют производственную «единицу». Азот в установках Клода получается из жидко го воздуха. Смесь исходных газов предварительно очищается, проходя через нагретую до 400° камеру с катализатором, в которой СО переходит в Н20 + СН4 (пары воды и метан). Последний используют далее в качестве топлива. 80—85% исходной газовой смеси переходит в NH3. В настоящее время по методу Клода строятся или уже построены в разных странах 14 предприятий с общей выработкой 97 и в день. 3) Английский метод, очень близкий к методу Габера—Боша, применяется на большом предприятии об-ва Brunner Mond, в Англии, которое вырабатывает пока 800 тонн связанного азота в год, а когда оно будет достроено, оно сможет вырабатывать до 50 000 тонн связанного азота в год. 4) Американский метод и
5) метод Фаузера имеют лишь второстепенное значение. Когда все строящиеся з-ды будут закончены, то ежегодное мировое производство синтетического А. по методу Габера — Боша и другим будет выражаться следующими цифрами:
Метод Габера-Боша.. .. 300 0 00 т
» английский. 50 000 »
» Клода. 30 000 »
Казале. 160 000 »
Фаузера. 10 000 »
» американский. 10 000 »
Всего. .. 560 000 т
Чрезвычайно важное значение в производстве синтетического А. имеет вопрос об исходных продуктах: водороде и азоте. Стоимость, непрерывность процесса и рентабельность метода находятся в большой зависимости от дешевизны, чистоты и доступности обоих газов, образующих синтетический А. Конечно, лучше всего было бы применять чистый электролитический водород (на 1 килограмм NH3 идет 2—2,2 j»t8 Н2), но получение электролитического водорода, во-первых, связано с значительными затратами основного капитала (по американ. вычислениям—до 400 000 долл, для постройки завода с производительной способностью в 28000 л3 Н2 в день), а во-вторых, и стоимость самой электриче. энергии для электролиза, при отсутствии дешевых гидро-электрич. ресурсов, довольно значительно отразится на цене синтетич. А. Поэтому электролитич. Н2 м. б. применен с выгодой лишь там, где он оказывается побочным продуктом при каком-нибудь другом электролитическом производстве, либо там, где он крайне дешев в виду мощности ресурсов «белого угля». На получение 1 м3 Н2(по методу Фаузера, Пехкранца, BAMAG и друг.) расходуется 5—6 kWh. Что касается азота, то его производство представляет лишь незначит. долю стоимости синтетического А. Его обычно добывают путем фракционировки жидкого воздуха (смотрите Воздух). При дешевом водороде чистый азот получается весьма оригинальным методом Китцингера. Дымовые газы, бедные содержанием кислорода и очищенные от пыли, смешивают и нагревают с генераторным газом в реторте, в присутствии катализаторов. При этом водород генераторного газа сжигается за счет кислорода, оставшегося еще в дымовых газах; после этого из образовавшейся от сжигания газовой смеси поглощаются пары воды, углекислота и другие газы, а азот в смеси с водородом в требуемой пропорции (1 : 3) поступает после очистки в контактную камеру габеров-ской установки. Сравнительнр дешевый Н2 выделяется из водяного газа (смотрите), добываемого при участии катализаторов при нормальном давлении и при t° в 500° согласно реакции: С0 + Н20=Н2+С02 (способ Боша). Процесс этот экзотермический и проходит без затраты топлива. Продукт, содержащий 30% С02 и ок. 2% СО, промывается под давлением в 25 atm водой, для удаления С02, и под давлением 200—225 atm—аммиачным раствором хлористой меди, для удаления СО. Водород добывается также по методу Казале из газа коксовальных печей (ок. 50% Н2, ок. 30% СН4, ок. 20% С2Н4, кроме того следы СО и N2) путем конденсации посторонних газов при помощи охлаждения (по методу Клода и Линде). На 1 ж8 Н2 требуется около 2Н Следует еще упомянуть о способе BAMAG и Пинса в Германии. Этот способ заключается в образовании водяного газа при действии паров воды на железо :2Fe + 3H20==ЗН2 + Fe203. Обратная регенерация Fe производится генераторным газом. На получение 1 ж3 Н2 расходуется около 3 килограмма пара и 1,3 килограмма кокса.
Наиболее выгодным методом получения сразу обоих исходных газов для синтеза А. является комбинированное применение водяного газа (2 объёма) в смеси с генераторным газом (1 объём). В водяном газе, путем введения в него избытка паров воды и предварительного пропускания его при 500° через железную контактную массу, равновесие СО + Н20 ^ С02+ Н2 сдвигается в сторону образования максимального количества С02 и Н2 (остается не более 1—2% СО). Коксогенераторный газ состоит из 30% СО и 70% N. Таким путем, после соответствующей очистки и обработки, можно сразу получить смесь азота и водорода в требуемой для синтеза NH3 пропорции (1 : 3); для этого к смеси нужно прибавлять лишь небольшое дополнительное количество чистого азота, добывая его по способу Линде из жидкого воздуха.
Цианамидный метод получения А. Хотя исторически метод фиксации атмосферного азота путем присоединения его к карбиду кальция с образованием цианамида кальция предшествует синтезу А. по методу Габера—Боша, все же цианамидный метод весьма скоро уступил пальму первенства га-беровскому методу; перспективы дальнейшего развития цианамидного метода ныне не обещают каких-либо значительных сокращений стоимости производства, которые могли бы вернуть ему преимущественную роль по сравнению с габеровским методом. Цианамидный способ фиксации азота (по идее Франка и Каро) основан на способности карбида кальция присоединять к себе атмосферный азот при t° около 1 000—
1100° с образованием цианамида кальция по реакции: СаС2 + N2=CaCN2 + С + +98 430 cal. Т. о. реакция образования этого препарата связанного азота является экзотермической и сопровождается выделением значительного количества тепла. Достаточно нагреть перемолотый в порошок карбид лишь для начала реакции до 1 000°, и дальнейший процесс присоединения N2идет самостоятельно, не только без затрат, но даже с выигрышем энергии. Зато получение исходного продукта карбида кальция связано с значительной затратой энергии на обжиг известняка (СаС03=СаО + +С02 — 42 900 cal) и на образование самого карбида (СаО + ЗС=СаС2 + СО — 121 000 cal). При образовании своем из карбида цианамид кальция сплавляется в массивные, весьма твердые барабаны. Имеются попытки сделать производство цианамида кальция непрерывным; например, этажные печи Карлсона (на шведских заводах Лыонга) или каналовые печи Польцениуса с перфорированными металлич. коробами, наполненными мелким карбидом и поступающими последовательно в длинную нагретую азотирующую печь (схему установки для азотирования карбида см. на фигуре 2).
| Фигура 2. Схема азотирова- | ния карбида по Франку и j | |||||
| Каро: А — перфорирован- | ный короб с молотым кар- | бидом, В — шамотовая азо- [ | С | ||||
| тирующая печь, С — угольный электрод, 1 см, D — i плотная крышка, К —приток | азота, F — другой электрод. J | А | f | |||
Присоединению азота к карбиду способствует прибавление к последнему хлористого кальция или, в особенности, плавикового шпата (фтористого кальция). В печах Карлсона перемолотый карбид, перемешиваемый автоматическими мешалками, подвергается действию встречной струи азота. Нагревание ведется при помощи зе-деберговских угольных электродов. На 1 тонна 12—20%-ного цианамида тратится 3,75— 5,50 килограмм электродного угля, причем цианамид получается уже не в виде массивных блоков, а в виде пористого материала, легко поддающегося перемалыванию.
Для получения А. из цианамида кальция последний обрабатывается в автоклавах под давлением в 11—12 atm парами воды. Предварительно в автоклавы вводится 3%-ный раствор NaOH. В течение 1г/2 часов разложение заканчивается, и NH3 м. б. либо конденсирован либо пущен в производство для получения тех или иных азотистых соединений (азотной кислоты, сернокислого аммония и т. д.). В связи с громадным успехом габеровского метода фиксации атмосферного азота, цианамидный метод вряд ли сможет в будущем успешно конкурировать с ним, но развитие циан амидной промышленности находится в тесной связи с производством карбида (смотрите) и с перспективами, стоящими перед утилизацией этого важного фабриката и получаемого из последнего ацетилена (смотрите).
В последнее время в германских установках пытаются использовать теплоту сгорания побочного продукта реакции, то есть газа СО (30% всей энергии карбидной печи), в качестве экономии энергии при фиксации азота по цианамидному способу; общая затрата энергии при связывании азота по цианамидному методу исчисляется в 13 000 kWh на 1 тонна связанного азота, то есть в 1js часть той энергии, которая тратится на связывание азота и при дуговом методе. Этим и объясняется большее развитие цианамидной азотной промышленности по сравнению с дуговым методом связывания атмосферного азота. Цианамид кальция может непосредственно применяться в качестве удобрения, но он служит также и для получения синтетической мочевины, гуанидина и др. концентрированных удобрительных туков, а также для получения цианистых соединений. При непосредственном применении в качестве удобрений цианамида кальция к нему приходится прибавлять немного воды (для разложения остатков непрореагировавшего карбида) и масла (иначе он очень сильно распыляется). Для получения А. из цианамида кальция последний обрабатывается при повышенном давлении водой. До синтеза Габера развитие цианамидной промышленности шло чрезвычайно бурно, в особенности во время войны, когда значительная часть А., полученного из цианамида должна была быть окисляема контактным методом в HN03. После войны развитие цианамидной промышленности заметно замедляется, уступая первое место габеров-скому методу (смотрите Азот). В настоящее время состояние цианамидной промышленности характеризуется следующими цифрами:
| Страна | Число пред прия тий | Годов, произв. способн. в т связ. азота | Продук ция
В 1926 г. в т связ. азота |
| Германия. | 4 | 90 000 | 86 000 |
| Франция. | 8 | 44 000 | 36 000 |
| Италия. | 4 | 18 000 | 1 8 000 |
| Норвегия. | 2 | 21 000 | 10 000 |
| Швеция. | 2 | 6 000 | 6 000 |
| Швейцария. | 1 | 3 000 | 3 000 |
| Польша. | 1 | 30 000 | 28 000 |
| Чехо-Словакия. | 1 | 5 000 | 3 000 |
| Юго-Славия. | 2 | 10 000 | 6 000 |
| Канада. | 1 | 18 000 | 18 000 |
| Япония. | 4 | 25 000 | 20 000 |
| С.-А. С. Ш. | 1 | 40 000 | Не работ. |
Следует отметить еще один метод получения синтетического аммиака, т. н. нитридный процесс (смотрите Нитриды). Так, например, при действии на окись алюминия азота в присутствии угля образуется нитрид алюминия, который водой разлагается с образованием NH3 по реакции: А1203 + ЗС + + N,= 2 A IN + ЗСО; 2A1N + 6Η20=2ΝΗ,+ +2ΑΊ(0Η)3. Но в виду целого ряда затруднений, с которыми связано производство NH-по этому методу, предложенному Серпеком (Soci6t6 g6n6rale de nitrures, D.R.P. 357 899), в смысле конструкции аппаратуры, этот метод не обещает в будущем больших успехов.
Статистические и промышленно-экономические данные о производстве синтетического А. — см. Азот.
Лит.: Кайзер К. пМозер А., Азот воздуха и его использование, М., 1922; Ост Г., Хим. технология, Л., 1924—27; W a ese г В., Die Luft-stickstoff-Inclustrie, Lpz., 1922; Partington J. R. and ParkerL, H., The Nitrogen Industry, L., 1922;
T. Э. m. I.
Grossmann H., Stickstoffindustrie u. Weltwirt-schaft, Stuttgart, 1926; Ammoniak u. Stickstoff, Fr. Ulhnann s Enzyklopiidie der techn. Chemie, B.— Wien, 1914 u. if.; Rideal and Taylor, The Catalysis in Theory and Practice, L. 1926; Pascal P., Synth6ses et catalyses industrielles, P., 1 925.