 > Техника, страница 95 > Аммиак
> Техника, страница 95 > Аммиак
Аммиак
Аммиак синтетический. Синтез А, из элементов в настоящее время является основным способом связывания атмосферного азота. Сырьем для этого процесса служит смесь азота и водорода в соотношении, б. или м. близко отвечающем составу N2+3H2. Помимо методов, применяющихся независимо от синтеза аммиака для производства азота и водорода, существуют специфич. методы производства такой азото-водородной смеси. Источником азота для синтеза А. является воздух, иногда даже после использования его кислородной части (дымовые газы, хвостовые нитрозные газы азотнокислотных установок). Источником водорода для установок большого масштаба является вода и некоторые водородсодержащие соединения в топливе, используемом для получения технологического газа. Процесс синтеза А. в заводском осуществлении состоит из следующих трех стадий: Л) производство первичного, или технологического, газа,
2) производство азото-водородной смеси, 3) синтез А. из элементов. Первые два этапа определяют собой около 75 % общей стоимости А., причем 60 % от общей стоимости падает в среднем на долю водорода. Таким образом выбор схемы производства водорода обычно решает вопрос о себестоимости продукции на заводе синтеза аммиака.
Получение первичного газа и азото-водородной смеси. Хотя сырьем для водорода в значительном количестве служит вода, но превращение ее в водород требует обработки, связанной со значительным, б. ч. бросовым, расходом энергии в виде топлива или гидра-влич. энергии; иногда носители энергии одновременно выступают как составная часть сырья; поэтому заводы синтеза аммиака географически располагаются обычно вблизи залежей ископаемого горючего, или источников нефти, или мощных водных потоков. Производство первичного газа может давать азот и водород в раздельном состоянии (что также пригодно для целей синтеза) или в виде определенной смеси, обычно с балластными и подлежащими удалению примесями. В первом случае водород может получаться: 1) электролизом воды, 2) разложением воды на железном катализаторе по обратимой реакции: 3Fe+4H20=Fe304 +4Н2; для реакции регенерации применяется водяной газ, получаемый газификацией кокса или крекингом метана:
Fe304 + C0=3Fe0 + C02;
Fe304 + 4Н2=3Fe + 4Н20;
3) в результате кислородной газификации угля или крекинга метана (азот присутствует в ничтожном количестве). Азот соответственно может получаться из воздуха или дымовых или нитроз-ных газов: 1) низкотемпературной обработкой, ректификацией жидкого воздуха или азото-кислородной смеси. после абсорбции высококипя-щих компонентов, 2) высокотемпературной обработкой, «выжиганием» кислорода и окиси азота в водороде, После абсорбции в случае необходимости таких составных частей, как С02. Окись азота раскисляется водородом на воду- и азот. Газификация кокса или угля и крекинг метансодержащих газов м. б. проведены так, что получающаяся газовая смесь после очистки представляет собой газ состава: ЗН2 : Ш2. Для этой цели в газогенераторном цехе подбирается режим паровоздушного дутья т. о., чтобы из генераторов выходил газ Примерного состава 37% Н2, 37% СО, 22% Na, прочее С02, H2S, либо перед газгольдером соответственно смешиваются водяной и воздушный газы обычно в пропорции 2,3 : 1 (при парокислородном дутье под уголь средний состав газовой смеси: Н2 40%, СО 28%, N2 4%, СН4 1,5%, С02 21%). Аналогично при конверсии метана согласно реакции СН4+Н20=С0+ЗН2 часть газа, которую необходимо сжигать в конвертере для поддержания баланса тепла, не выбрасывается в атмосферу, а дозируется в качестве добавки к основному технологическому газу. В результате получается состав газа с необходимым содержанием азота (52% Н2, 22% СО, 24^% N2). Подобный газ однако представляет собой лишь первичный газ для синтеза А. Он подлежит перёработке с целью удаления соединений, являющихся ядами или балластом для реакции синтеза аммиака из элементов; в качестве ядов особого внимания требуют кислородные (СО, С02, Н20, NO) соединения и сам кислород, сернистые (H2S, С32, COS) и некоторые высокомолекулярные органические соединения (С10Н8, С6Н6); эти соединения или непосредственно выделяются из газовой смеси или перерабатываются в полезные продукты; в качестве балласта т. н. инертных газов фигурируют метан, аргон. Они могут присутствовать при реакции синтеза аммиака в значительно большем количестве, чем яды, но эффект разбавления реагентов сказывается на реакции также отрицательно. Содержание кислородных соединений в очищенном газе в ряде методов достигает цифры 5-10~6то есть 0,0005% [*].
Переработка СО производится обыкновенно согласно реакции
СО + Н20=С02 + Н2 + 10 800 Cal на железном катализаторе с хромовым активатором при t° ок. 500°. Смесь водяного и воздушного газов превращается например в газ состава 51% Н2, 17% Nj, 30% С02, 2% СО. Этот процесс проводят в пром-сти при обычном давлении и под давлением до 30 atm ведутся опыты по конверсии СО водой в жидкой фазе в условиях, близких к критическому состоянию воды. (В настоящее время ведутся опыты доломитной конверсии и с низкотемпературным катализатором, например кобальтом.) Очистка газа от сероводорода обычно происходит в процессе специальной предварительной очистки, абсорбирующей H2S железо-содовым раствором (метод Фер-рокс), содово- или аммиачно-овокислым (метод Тайлокс) или же железорудным отвалом Fe203 (сухой метод) с последующим полным (Тайлокс) или частичным использованием серы сероводорода, выделяемой в элементарном состоянии. Следы сероводорода поглощаются в следующих ступенях очистки, состоящих из промывания газа водой под давлением (удаление С02, H2S), раствором щелочи, примерно 10%-ным (следы С02), раствором А. под давлением (С02>HaS), аммиачно-медным раствором уксусной или муравьиной кислоты (СО, 02). Давление, в весьма значительной степени увеличивая эффективность процесса, легко достижимо» поскольку синтез требует давления 100—1 000 aim, а очистка водой Ί2—30 ат такое же давление достаточно для А. и щелочи; купро-аммониевый комплекс требует 100—200 atm. Очистка водой м. б. заменена очисткой аммиачным раствором и, наоборот, в зависимости от водных ресурсов места постройки завода и целесообразности устройства оборота в водном хоз-ве. Современные масштабы синтеза А. вызывают огромные расходы проточной воды, что позволяет применять такую схему лишь у полноводных рек. Например завод производительностью 30 000 тонн NH3 в год (мощности, ниже которой не советуют идти немецкие экономисты) потребляет в час летом ок. 3 500 тонн воды, а вся Москва ок. 20 000 том числе При некоторых условиях, например при наличии в комбинате завода сульфата аммония по гипсовому методу, целесообразно даже включать последовательно водную и аммиачную промывки в соответствующей пропорции. Помимо метода очистки абсорбцией широко распространен т. наз. метод глубокого охлаждения, состоящий в разделении компонентов газовых смесей путем фракционированной конденсации. В газовой фазе в конечном счете остается Н2и небольшое количество азота, а также следы СО. Подобным образом можно конечно изготовить и чистый водород, причем происхождение первич ного газа м. б. весьма различным: обычный коксовый газ, водяной газ, отходящий газ пиролиза нефти, газ парокислородной газификации. угля после конверсии ит.п.; При изготовлении чистого водорода требуется большая теплообменная поверхность и следовательно большие металловложения, поскольку здесь к эффекту специального хладоагента не присоединяется полезный эффект расширения сжатого (дозируемого для смеси 3H2:1N2) азота. Обычно при подобном способе выделения водорода азот получается ректификацией жидкого воздуха, но непосредственной технологической зависимости между обоими процессами нет. Ректификация воздуха целесообразна например при одновременном использовании кислорода в дутье под генератор. Преимущество мзтода «вымораживания» (глубокого охлаждения) заключается в обеспечении удаления метана из газовой смеси, чего в техническом масштабе путем абсорбции достигнуть пока не удается. Кроме абсорбции и конденсации иногда приходится прибегать еще к очистке газа методом конверсии. Замысел этого метода заключается в превращении вредных соединений в инертные, безвредные для катализатора примеси или в побочные продукты (та же идея по существу заложена в конверсии СО в Н20 и С02, но размеры ее заставляют выделить эту операцию в специфич. процесс). Такнапример, по патенту Клода между цехом глубокого охлаждения и цехом синтеза вырабатывается метанол из остаточного СО и избыточного Н2 по реакции: СО + 2Н2==СН3ОН 4* 53 000 Са1 в присутствии хромовоцинкового катализатора. В большинстве случаев на современных з-дах, учитывая незначительное содержание СО в газе перед синтезом (0,01—0,001%), упрощают процесс, превращая СО в метан по реакции
СО + ЗН2=Н20 + СН4 + 48 000 Са1.
Этот процесс м. б. совмещен с производством А. и производиться в идентичных аппаратах в присутствии идентичных катализаторов при условии обеспечения термин, режима, то есть нек-рого подвода тепла во время реакции. В таких случаях подобный предварительный катализатор, т. н. форконтакт, естественно довольно быстро теряет активность, подвергаясь отравлению в первую очередь; срок его жизни, то есть продолжительность действия, зависит от содержания каталитических ядов в газе (кислородные, сернистые, фосфорные соединения); зато введением фор-контакта обеспечивается плавная и более длительная работа основных агрегатов синтеза.
Среди не получивших еще широкого промышленного развития, но многообещающих процессов можно отметить: 1) форконтакт селективного характера, превращающий СО в присутствии Н2в С02 (гопкалиты), 2) для маловодных местностей с паром высокой стоимости, делающей невыгодной диссоциацию карбоната аммония на А., замена обычной промывки от СО2 на этано-ламиновую промывку (смотрите Этаноламины), 3) абсорбция метана бензинами при — 35°, что обещает значительно сократить расход энергии сравнительно с методом глубокого охлаждения. Наконец уже осуществлены в форме опытных агрегатов: а) парокислородное дутье под низкосортный уголь, исключающее процесс коксования из хозяйства синтеза и уменьшающее потребность в коксе в стране, газификация лигнитов во взвешенном состоянии [а], б) конверсия СО непосредственно в домне или получение первичного газа для синтеза А. в домне и др. Ниже показано распределение методов производства водорода для азото-водородной смеси на земном шаре на 1934 г. (в %) [V]:
Электролиз воды.. 12,9
Глубокое охлаждение коксов, газа. 24 О
Водяной газ .. 59,8
Крекинг природных газов. 0,8
Прочие методы (железопаровой способ, газы брожения и др.). 2,5
Азот за исключением нескольких небольших установок (Мерано в Италии — крекинг нитроз-ных газов; Слюйскил в Голландии — ректификация смеси воздуха и топочных газов; Найа-гара Фоллс в США — выжигание кислорода воздуха водородом) готовится разгонкой жидкого воздуха. Характеристика различных способов производства азото-водородной смеси по фактическим расходам на 1 mNH3 (2 850 м3 3Η2+Ν2) приведена в таблице 1.
Эквиваленты: 1 kWh=0,80 килограмм У. Т. (условное Топливо); тепловой эквивалент 1 килограмм У. Т.— 7 000 Gal; для получения 1 kWh электроэнергии на современных электростанциях тратится 0,8 килограмм У. Т. (полагаем кпд электростанций 15%), 1 тонна пара среднего давления—120 «а
Таблица 1. — Характеристика способов производства азотоводородной смеси.
| Способ производства | Энергия в kWh | ε
η Si |
Пар в m | Кокс в m | η
ей и d „ о ^ Й 02 |
•«
S “ Л |
Антрацит т | Отход богатого газа в м“ |
| 1. Электролиз воды + перегонка воздуха .. | 12 840 | 120 | ||||||
| 2. Глуб. охл.: конверсия газа + перегонка воздуха. | 2 070 | 810 | 0,2 | _ | 4 650 | _ | _ | 2 510 |
| 3. Водяной газ + конверсия СО | 727 | 955 | 10,0 | 2,1 | — | — | — | — |
| 4. Крекинг натур, газов + конверсия СО.. | 657 | 730 | 10,0 | _ | _ | 1 345 | _ | __ |
| 5. Железопаровой процесс + перегонка воздуха.. | 1 330 | 310 | 11,1 | - | - | - | 2,9 | - |
Фигура 2.
ВТ
| р | -00-
if |
"Н----(.----п
! · « ; » ill 1 1 1 1 i 1 J |
ZZXcoy
19 i | |||
| !
=? 1 |
1
1 |
ifrt-fft, Л ! Μ | -~Ί
1 1 L _га. |
rb.
tfc:
Условные обозначения
----Вода
---Щелочь
— - Богатый газ
— - — Окись углерода --Металл
—— Воздух высок, давл —Азот высок давл
-----Кислород
----Азот низк. давл
----воздух казн, давл
-Аммиак
-Азота -водород» смесь
Коксовый газ
Фигура 2.
У. Т., 1 ООО мг воды в комбинате —100 килограмм У. Т., 1 м3 коксовального газа 3 820 Gal, 1 mz естественного газа 7 400 Gal, 1 м3 богатого газа 4 900 Gal [2]. Расчет дан для 2 850 м3 ЗН2 -{- N2(при нормальных условиях), сжатых до 300 atm. В способах 2, 3, 4 очистка от С02 принята водная, в способах 3,4 — паровой привод в компрессии. Ниже дана энергетич. характеристика различных способов получения азото-водородной смеси (в кг У. Т. на 1 тонна NH3).
| Способы | Энергия в виде тока | Воды | Пара | Горю чего | ОТХОД | Сумма |
| 1. | 10 300 | 12 | _ | _ | _ | 10 312 |
| 2. | 1 640 | 81 | 24 | 2 540 | 1 755 | 2 530 |
| 3. | 580 | 96 | 1200 | 1 720 | — | 3 596 |
| 4- | 530 | 73 | 1 200 | 1420 | — | 3 22В |
| 5. | 1060 | 31 | 1 450 | 2 708 | — | 5 249 |
| Соотношение между | азотом | и водородом ил- | ||||
люстрируется таблицей 2.
Таблица 2. —Соотношение между азотом и водородом при различных способах производства.
| Способ производства азота | Энергия в kWh *1 | Вода
В М**1 |
Водород
В М* |
| Ректификация воздуха. | 420 | 45 | _ |
| Крекинг нитрозных га | |||
| зов .. | 236 | 63 | 51,5 ** |
*1 На lm NH3 (700 мз N2 при нормальных условиях сжатого до 300 aim). *2 при переводе водорода на энергетич. эквивалент общий расход энергия достигает 513 kWh.
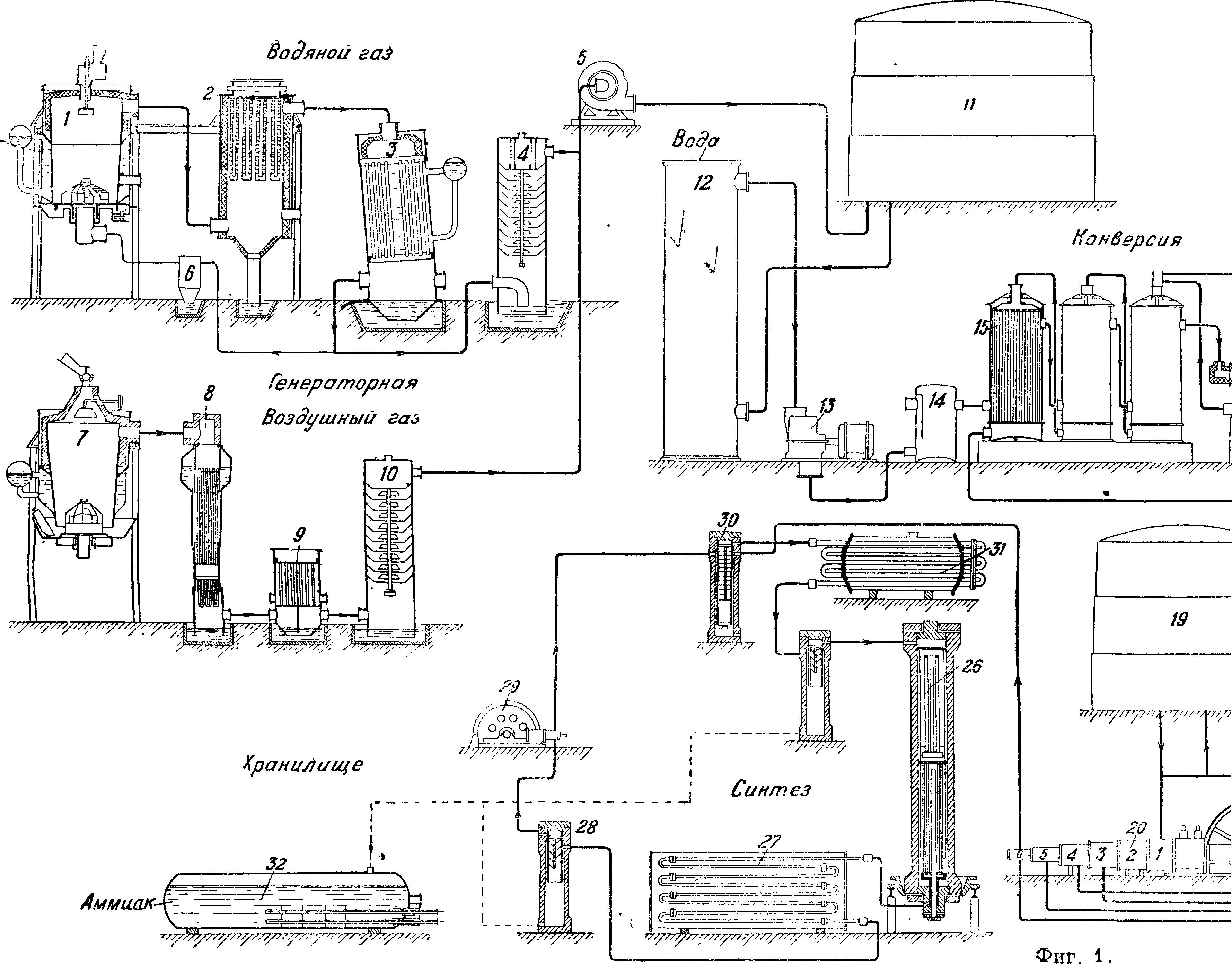
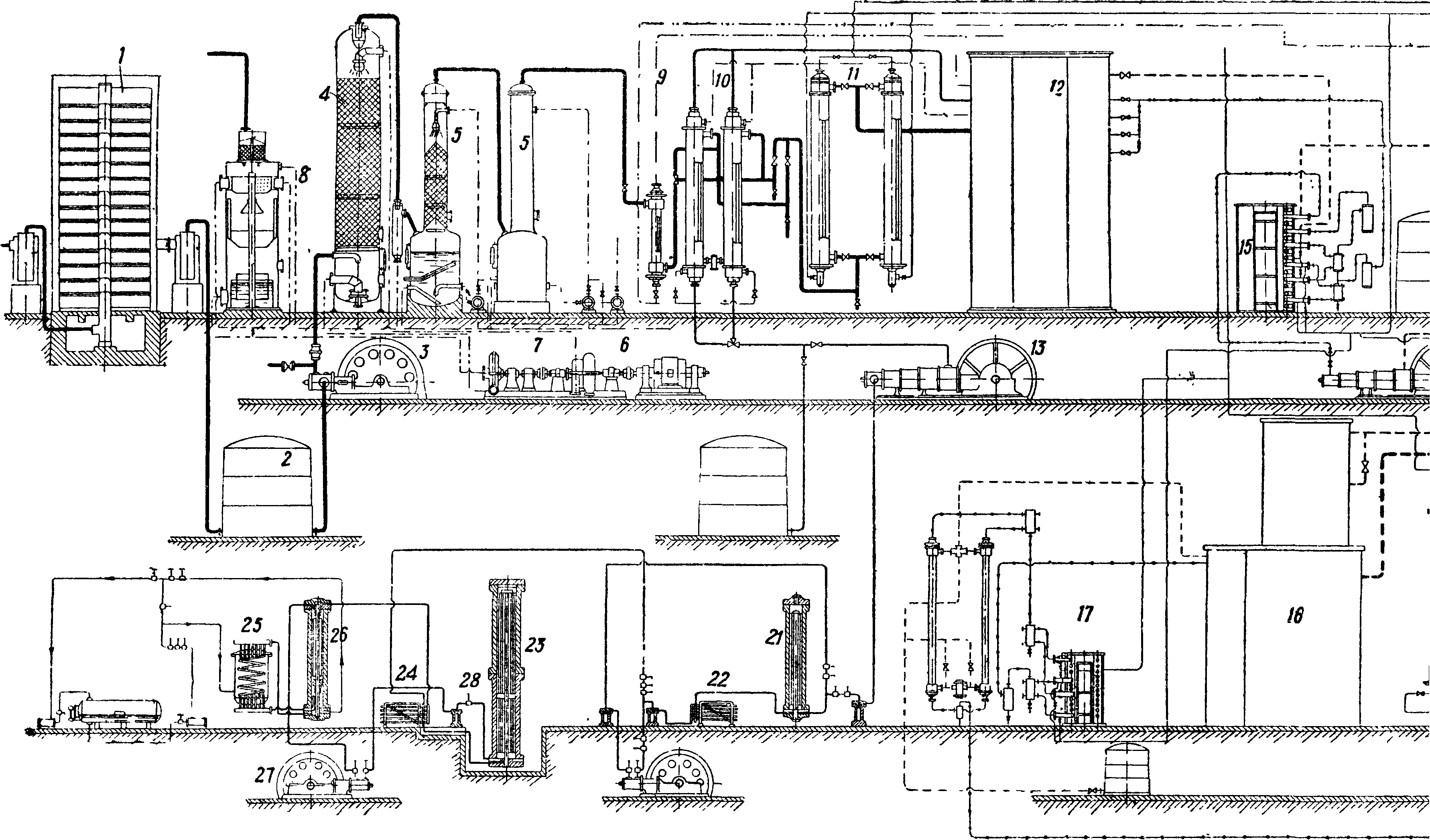
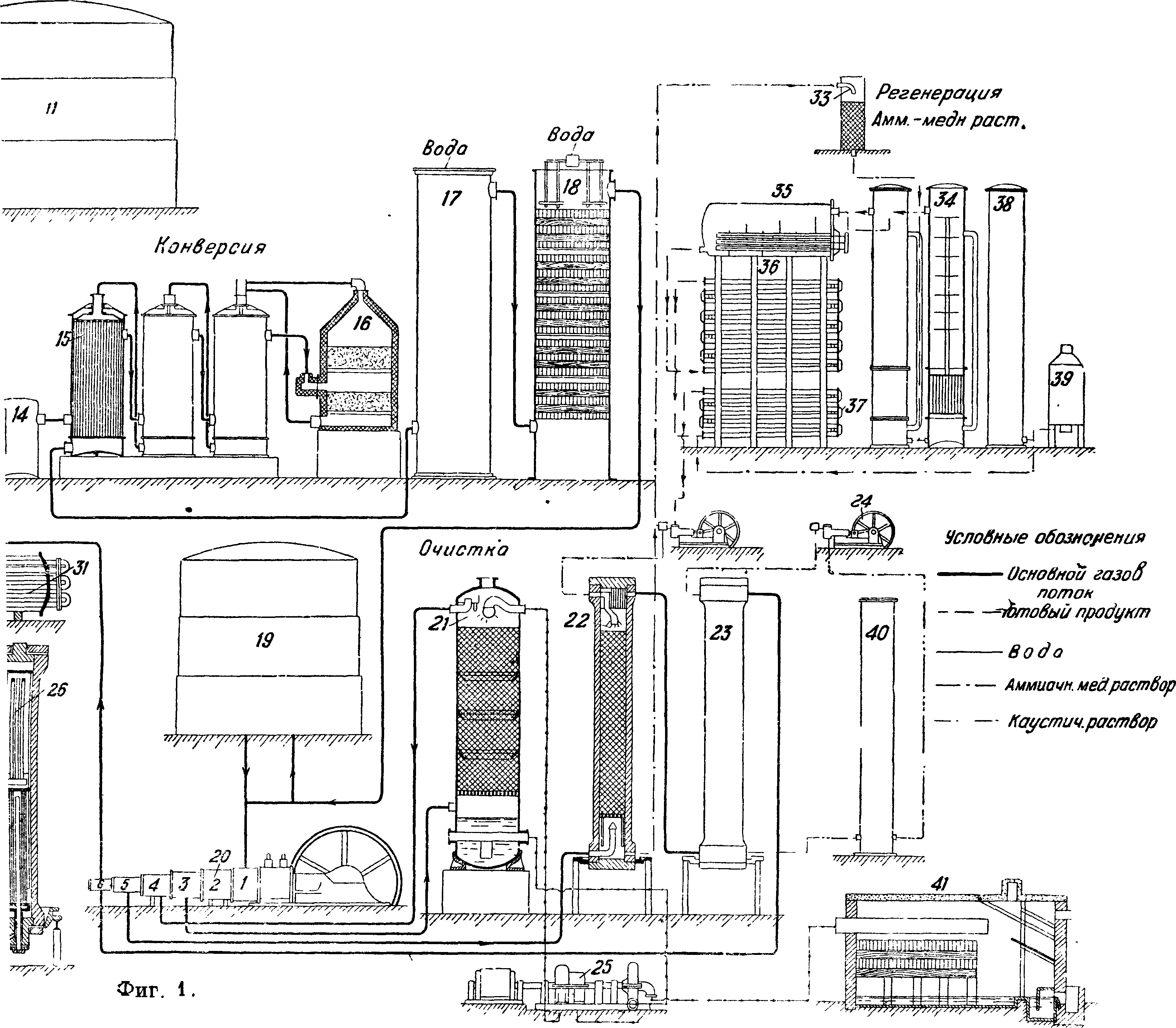
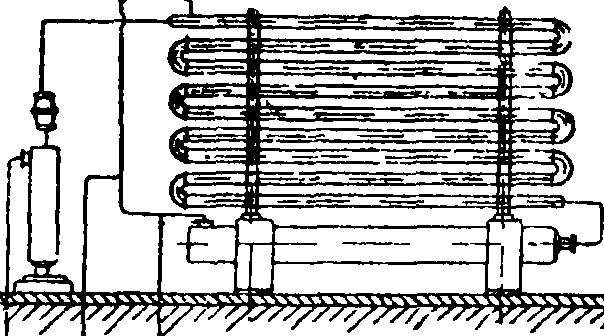
Очищенная азото-водородная смесь поступает в цех синтеза. В производстве приготовление этой смеси и синтез аммиака являются неразрывными по существу звеньями единого комплекса, хотя и комбинируются в различных сочетаниях. Номенклатура заводов синтеза привилась следующая: Линде-Фаузер, Клод-Клод и др. Здесь первое имя относится к автору схемы производства водорода, второе — к автору метода синтеза А. из элементов. Если встречается лишь одно слово, например «Найтроджен», «Казале», то это значит, что обе стадии производства разработаны одним изобретателем или принадлежат одной фирме (фигура 1 и 2).
На фигуре 1 показана примерная схема производства А. Производство первичного технологического газа: в генераторе 1 при дутье водяного пара получается водяной газ, к-рый проходит рекуператор с насадкой 2 и котел-утилизатор 3, где, охлаждаясь, продуцирует пар. Далее газ поступает в промыватель 4 и газо-дувку .5; перед газодувкой к водяному газу добавляется воздушный газ, полученный из специального генератора 7, охлажденный в котле-утилизаторе 8, теплообменнике 9 и скруббере 10. Пыль оседает в пыльнике 6. Получение азото-водородной смеси из первичного газа. Из газгольдера 11 газ поступает в сатуратор 12, где насыщается водяным паром при темп-ре 85° посредством орошения горячей водой, затем в газодувку 13, в паросмеситель 14 и, пройдя теплообменник 15, в конвертер окиси углерода 16. Обогатившись водородом, газ проходит теплообменник в обратном порядке, затем охлаждается дальше в водонагревательном и конденсационном скрубберах 17 и 18. Горячая вода из скруббера 17 вместе с конденсатом избыточного пара поступает в сатуратор 12, чем экономится расход свежего пара. Конвертированный охлажденный газ направляется в газгольдер 19 или поступает в компрессор 20, откуда направляется на очистку от углекислоты 21 и окиси углерода 22 и в контрольный скруббер со щелочью 23. Чистый газ дожимается до конечного давления и подается в агрегаты синтеза (применяется давление от 100 до 1 000 atm; предпочтительно 800—1 000 atm). На фигуре 1 показан цикл воды для очистки газа от углекислоты: насос 25, спаренный с турбиной, регенерирующей часть энергии воды, и экспансер 41, где выделяется после дросселирования основная часть растворившихся газов. Также показан цикл очистительной жидкости для вымывания окиси углерода: холодный раствор из 22 дросселируется, поступает в редуцеры 34, затем в регенератор 35, где раствор подогревается паром, окись углерода выделяется и отправляется в цех конверсии СО для использования; увлеченный ей раствор улавливается в водных абсорберах 33. Регенерированный раствор охлаждается в водяных 36 и аммиачных 37 холодильниках, снова сжимается в специальных насосах 24 и снова поступает на орошение скрубберов высокого (120 atm) давления. Установка имеет бак для приготовления 39 и хранения 38 активного раствора, получаемого погружением меди в уксусную к-ту. Щелочь для орошения скруббера 23 хранится в баке 40 и подается на скруббер 23 с помощью насоса 24 с давлением 420 atm. Синтез А. из элементов. Основная реакция происходит в колонне синтеза 26, имеющей вверху катализаторную зону и внизу теплообменник; горячий газ по выходе из колонны охлаждается в водяном конденсаторе 27 (выделившийся А. сепарируется в 28), проходит циркуляционную помпу 29, масляный фильтр 30, конденсатор 31, охлаждаемый кипящим А., сепаратор к нему и снова возвращается в колонну. Свежий газ подается перед фильтром. Из сепараторов жидкий аммиак отправляется в сборник 32.
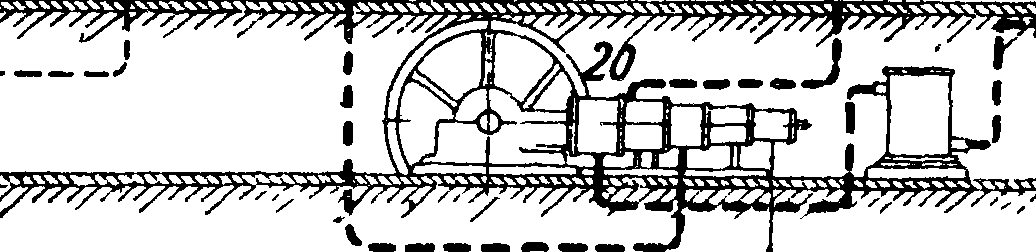
На фигуре 2 показана другая примерная, схема производства А. 4. Производство первичного га-з а. Первичный технологич. газ, коксовый газ, получается с коксовален после удаления основной массы бензола, нафталина, аммиака и смол. 2.Получение азот о-в о-дородной смеси. Коксовый газ освобождается от серы в башне 1, загруженной железной рудой, и поступает в газгольдер 2, откуда направляется в компрессор 3 с давлением выхлопа 12 atm. Сжатый газ промывается водой в скруббере 4, затем щелочью в δ и направляется в «блок разделения» коксового газа. Вода подается насосом 6; ее энергия рекуперируется водяной турбиной 7. Газы, растворенные в воде, выделяются в экспансере 8. Фракционная конденсация начинается в теплообменниках предварительного охлаждения 9 и 10, затем газ проходит аммиачные холодильники 11, откуда с темп-рой —45° поступает в кабины глубокого охлаждения 12, где при темп-ре —490° газ промывается жидким азотом, растворяющим окись углерода. Из кабин выходит почти чистая смесь ЗН2 + 1Ν2 со следами окиси углерода; эта смесь проходит в обратном коксовому газу порядке 11, 10 и 9 и идет в компрессор 13, где сжимается до давления синтеза А. (800—300 atm). Источником холода служит расширение азота, к-рый сжимается в специальном компрессоре 14 до 200 atm, затем охлаждается в аммиачных холодильниках 15 и, дросселируясь в кабинах глубокого охлаждения обеспечивает получение темп-ры (—190°), при которой под давлением 12 atm не сжижается лишь водород. Часть азота расходуется на растворение окиси углерода, часть поступает, пройдя специальный газгольдер, снова в азотный компрессор. До кабин глубокого охлаждения газ разделяется на метан и богатый (калорийный) газ, после кабин получается азото-водородная смесь и смесь N2 + СО (т. н. бедный газ). Чистый азот может получаться различными путями; обычно его получают вместе с кислородом ректификацией жидкого воздуха в установке, состоящей из ректификационной колонны 16, предварительного охлаждения воздуха высокого и низкого давлений 17, 18, щелочных скрубберов 19 и воздушного компрессора 20. 3.Синтез А. из элементов. В фбрконтакте 21 следы СО гидрируются в метан и воду; вода выделяется в холодильнике 22; свежий газ смешивается с горячим газом из колонны синтеза (пройдя ее внутренний теплообмен) 23 и идет на конденсацию в конденсаторах водяном 24 и аммиачном 25 и холодообменнике 26; в последнем происходит сепарация; затем газ проходит циркуляционную помпу 27, маслоотделитель 28 и снова идет в колонну синтеза 23.
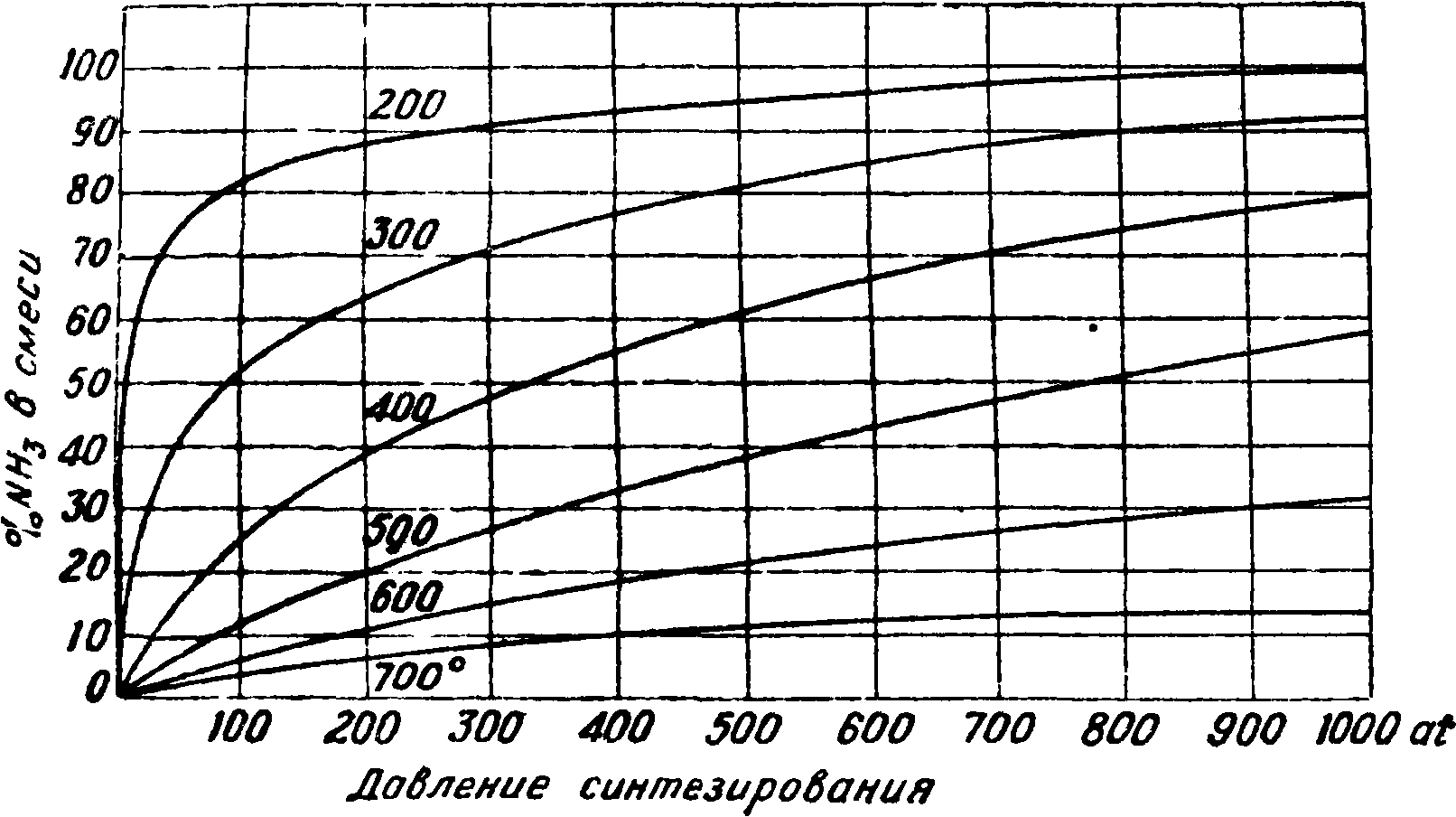
Синтез А. из элементов. Единой чертой для всех производственных процессов синтеза аммиака из элементов является сочетание следующих условий: 1) давление в аппарате синтеза в пределах 100—1 000 atm [5], причем в технике синтеза давления до 200 atm считаются низкими, до 600 — средними и лишь выше 600 atm — высокими. Давление создается многоступенчатыми поршневыми компрессорами, причем иногда последние одна или две ступени (при отношении компрессии, равной 3—2,5) выделяются в отдельный т. н. гиперкомпрессор по соображениям конструкции и обслуживания; 2) наличие катализатора в агрегате синтеза, в настоящее время в основе своей повсюду состоящего (до загрузки) из Fe304, с различными активирующими добавками в количестве 1—4%; активаторами служат щелочные или щелочноземельные окислы (К, Са) плюс амфотерные окислы (А1, Сг). Промышленную скорость процесса синтеза подобные катализаторы обеспечивают лишь при t° 450—650°, что для реакции N2+3H2=2NH3 +24 000 Gal приводит к равновесным выходам (фигура 3).
Фигура з.
Промышленная скорость синтеза соответствует 10 000—50 000 объёма газа в час при нормальных условиях на 1 объём (брутто) катализатора, причем выход достигает 80—70 % от равновесных величин, приведенных в таблице 3 (смотрите также фигура 4).
Таблица З.-Содержание А. в газовой смеси в % при равновесии.
| Температура в °С | Давление в atm | ||||
| 1 | 100 | 300 | 600 | 1 000 | |
| 0 | ί 9,60 | 98,90 | 99,35 | 99,59 | 99,64 |
| 500 | 0,13 | 10,40 | 23,10 | 42,15 | 57,50 |
| 600 | 0,05 | 4,50 | 11,50 | 23,10 | 31,43 |
| 700 | 0,02 | 2,18 | 7,28 | 12,60 | 12,87 |
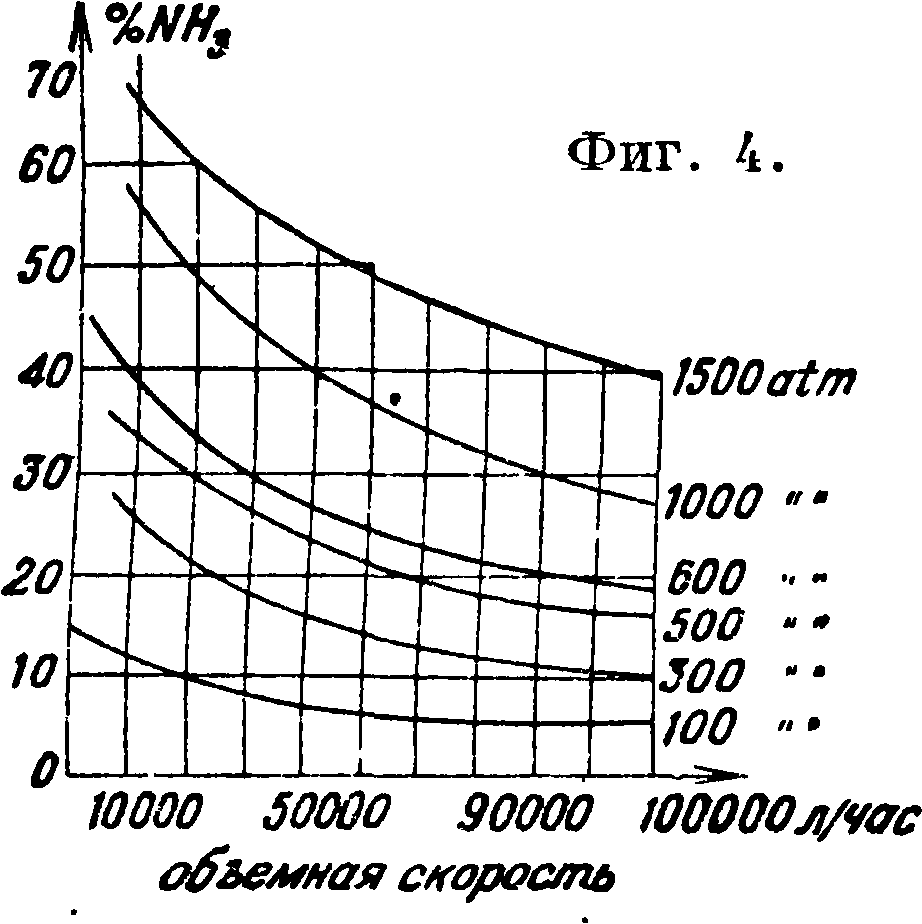
В современных конвертерах загружается от 0,5 до 2,5 м3 катализатора. Производительность его зависит от средней г°, которая в свою очередь определяется конструкцией колонн синтеза. Ведутся опыты, направленные к понижению рабочей t° для катализатора (применение нитрида лития и тому подобное.). Железный катализатор изготовляется из стали в кислородном пламени или переплавкой естественного магнетита с добавками (KN03,A1203 и др.). 3) Устройство системы синтеза, состоящей из ряда последовательно расположенных контактных аппаратов (Клод) или образующей замкнутый т. наз. цикл синтеза (прочие системы), вызвано сравнительно малой степенью конверсии, то есть неполным превращением исходной смеси в А. за один проход аппарата. Обязательными элементами цикла синтеза являются: а) конвертер, б) выделение А., которое раньше производилось вымыванием водой, сейчас же — исключительно путем конденсации А. в один или несколько приемов с применением естественного и искусственного охлаждения, в) циркуляционный насос для проталкивания смеси в цикле; функции насоса может исполнять инжектор, работающий от компрессора, восполняющий потерю давления в аппаратах, поскольку сам синтез идет под постоянным давлением, г) подвод свежей смеси и д) отвод продувки. Продувка является средством отвода из цикла балластных примесей (Аг, СН4), не успевших раствориться в жидком А. Эти примеси влияют на процесс синтеза согласно ф-ле
--__= К —Г1’5 Μ _ 1)2. р
(1 — Я)2 (1 +г)2 Н Г
(а — доля NH3 в равновесии, К— константа равновесия, г=-5L; i—доля инертных веществ,
Р—общее давление), то есть примеси понижают рабочее давление пропорционально квадрату содержания инертных веществ. Циркуляция и продувка в системе Клода отпадают.
Производительность конвертера П легко вычислить по следующей общей формуле, справедливой при различных схемах цикла
П=В
Кг-К2
100 + кг
где В — объём (в м3) циркулирующего газа, входящего в колонну; (Р, Ткатализа) — %
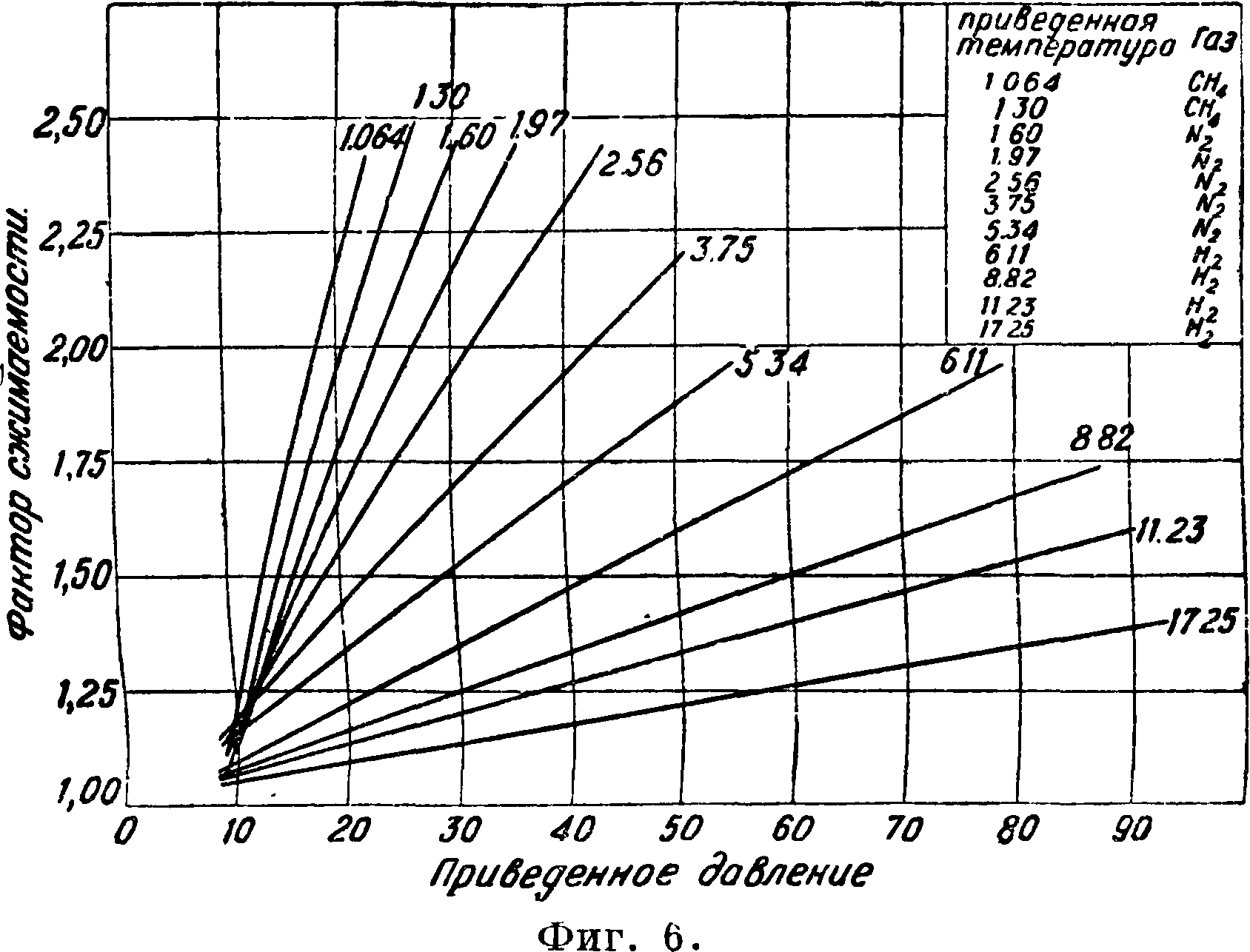
(объёмный) NH3 на выходе из колонны; К2 — — {Р, Тконденсации)—% (объёмный) NH3 на входе в колонну. Для пуска колонны синтеза необходим подогрев внешний или внутренний; при установившемся режиме процесса в хороших конструкциях работа колонны автотермична; в некоторых старых аппаратах приходится непрерывно подогревать входящую смесь (обычно электрич. сопротивлением). Обычные газовые показатели, как ур-ие состояния, закон Дальтона, Ср, Кр, практически при высоких давлениях неприложимы. Теория дает весьма громоздкие ф-лы для этого случая, и в конечном счете предпочтительно пользоваться графиками, построенными на основе эмпирич. данных. Небезынтересно лишь указать общий вид этих ф-л: 1) Взамен ур-ия Клапейрона следует применять выражение βΡν—RT, где β — коэф. сжимаемости, являющийся ф-ией давления и темп-ры (фигура 6).
Температура С°
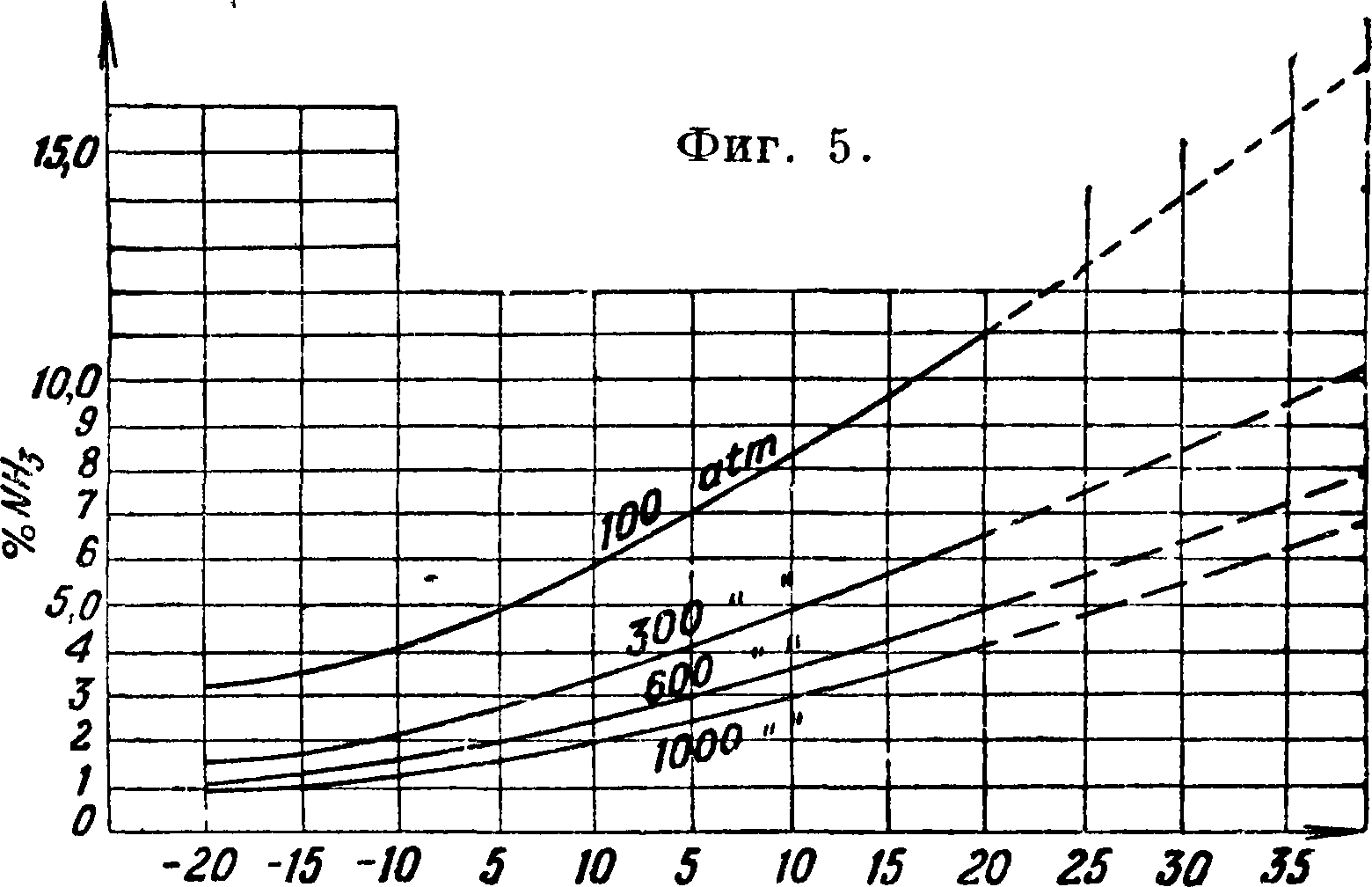
Для сравнения коэф-тов различных веществ (Ν2, Н2, ΝΗ3) на оси абсцисс (фигура 6) отложено приведенное давление, то есть отношение давления к критик. давлению. 2) Парциальное давление насыщенных паров как ф-ия t° в условиях высокого давления и динамич. потока значительно выше обычных цифр. Так, для давления 300 atm поправочный коэф. равен 2—2,5 (фигура 5). Кроме того закон Дальтона, основанный на аддитивности объёмов и давлений, здесь недействителен и должен быть заменен соотношениями, выраженными формулой [6]
P=RT [/,(Р„ V!, Χύ + ЫР; У; *.)+···].
где×— мольные фракции компонентов. Для смеси 3H2+N2 поправка достигает +1,5% при
0°, уменьшаясь с повышением г°. 3) Теплоемкость Съ можно вычислить по ур-ию
Р
р dW
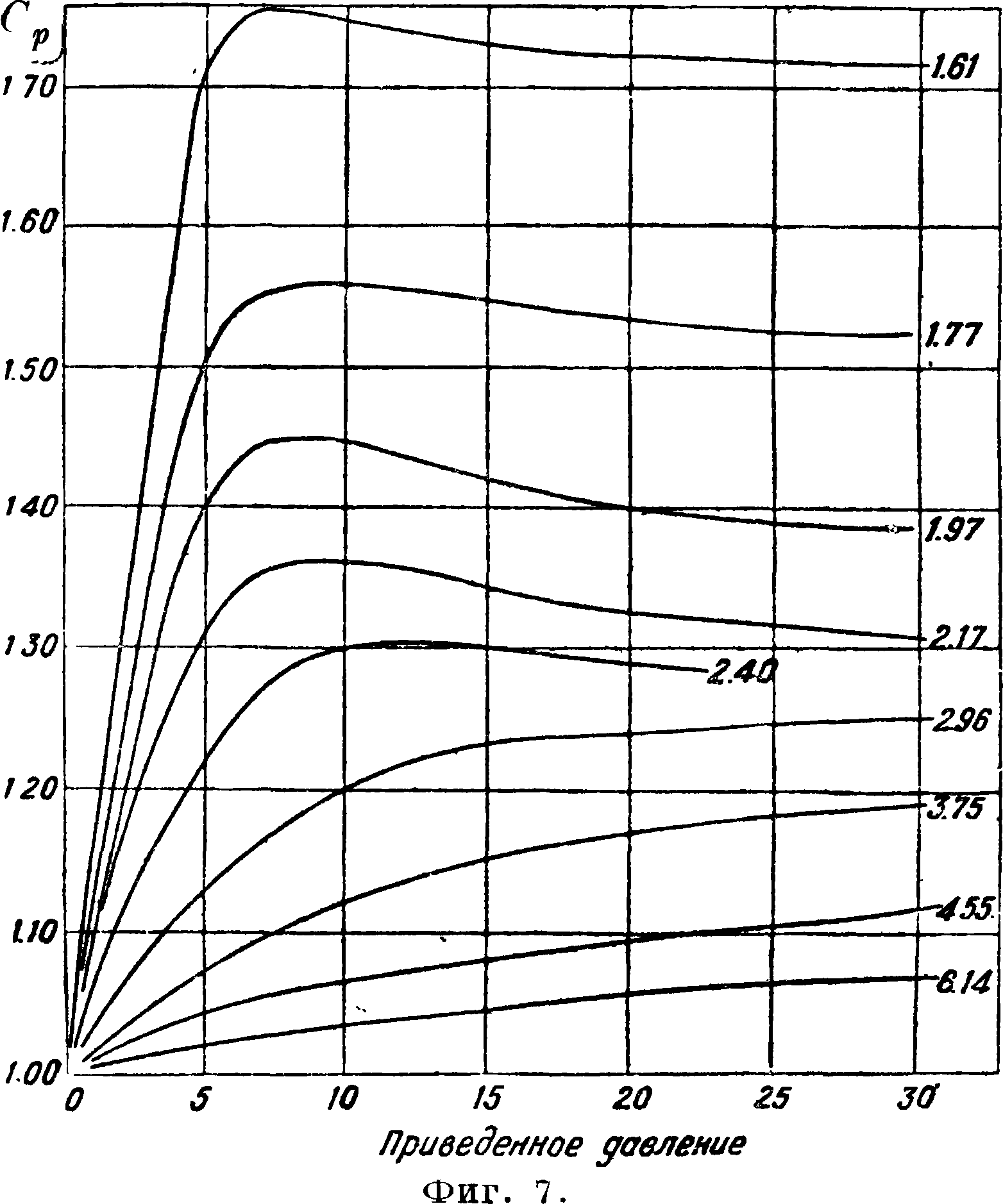
СР=СР0 + Т j cm PdP-Ро но вычисления по ур-иям состояния настолько хлопотливы, например по Кийсу
RT А
Р а (У— о2’
ν-(£Γν
что хотя они до 200 atm дают ошибку лишь ~0,5% при применении опытных постоянных,
удобнее пользоваться графиком (фигура 7). Последним надо пользоваться с осторожностью при вычислении Ср смеси. Напр. смесь 3H2+-N2 при давлении 300 atm имеет Ср около 1,0; если же полагать теплоемкость смеси величиной аддитивной, то из значений Ср для N2и Н2 для смеси получается значение Ср~ 0,85. 4) Крдля газовых смесей под давлением для соблюдения точности следует изобра-
аить KPo=Kp—f(P’ но отклонение составляет менее 1% от значения константы, поэтому им можно пренебречь. 5) В присутствии инертных веществ в практич. условиях оптимум реакции лежит при соотношении
Н2 _ о
N2 “
Поведение металла в условиях давления и в атмосфере водорода изучалось с различных точек зрения, но в стройную концепцию все еще не объединено. Простая углеродистая сталь разрушается в условиях синтеза с образованием пузырьков метана. Надо различать условия работы корпуса конвертера, испытывающего высокое внутреннее давление и t° порядка 0—100° (изредка 300°), и условия для внутренних частей конвертера, выполняющих функции теплообменника и носителя катализатора; для внутренних частей здесь перепад давления составляет около 2—20 atm, но t° достигает 500—600°. Кожух делают из хромованадиевой или хромоникелевой стали, допускающей рабочие напряжения до
1 080 килограмм /см2 (Найтроджен) или до 2 200 килограмм/см2 (Казале). Внутренние части становятся хрупкими и разрушаются в течение нескольких месяцев, даже изготовленные из специальной стали; поэтому их иногда конструируют из простой углеродистой стали. В табл. 4 приводится сравнение распространенных систем синтеза по основным показателям (продукция: жидкий А. 98—99%, прочее—вода и растворенные газы). Расход энергии на сжатие вычислен для очищенной
Таблица 4. — Сравнительные примерные характеристики различных систем синтеза А.
| Показатели | Клод | Казале | Фаузер | Габер-Бош
(Найтрод жен) |
удэ |
| Давление в aim. | 950-1 050 | 550-850 | 200-300 | 200-300 | 90—120 |
| Схема. | Открытая | Свежий | Помпа-> ко | Свежий | Свежий |
| цепь; в новых | газ —>· пом | лонна;
свежий |
газ -» пом | газ -► пом | |
| установках | па -> ко | па -*конден | па — ко | ||
| замыкается, | лонна; | газ -> кон | сатор -► ко | лонна -> | |
| превращается | конденса | денсатор -> | лонна; | конденса | |
| в цикл | тор отдув-ка | отдувка (фигура 1) | конденсатор -* отдувка (фигура 2) | тор | |
| Агрегат. | Блок 5 колонн | 1 колонна | 1 колонна | 1 колонна | 2 колонны |
| Диаметр (внутр.) | с конденсаторами дает 60 m NH3в сутки | до 80 m | 30—50 m | до 80 m | по 25 m |
| единицы в миллиметров | 2-380+3.240 | 400-600 | 850 | 700-1 000 | 1 000 |
| Состав газа (%NH3): на выходе из | 15-18 | 10-8 | |||
| колонны. | 25,0 | 20-25 | 10 | ||
| на входе *. | В блок - 0; на входе в прочие 3,0-0,5 | 2-8 | 2 | 2 — 4 | 0,5 |
| Объемная ско | 12 000—25 000 | 3 500—5 000 | |||
| рость в аппарате | До 50 000 | 30 000—45 000 | 15 000—20 000 | ||
| Расход энергии: | 230 | 212 | 100 | 400 | |
| а) без сжатия | — | ||||
| б) со сжатием Распростран.в % | 1 550 | 1 510 | 1 124 | 1 100 | 1 300 |
| на земном шаре | 10,7 | 45,6+8,2 | 8,0 | ||
| на 1934 г. | 9,3 | 16,7 |
азото-водородной смеси. В тех случаях, когда данный метод синтеза комбинируется со способами, очищающими газ При 12 aim или 16,0 (например Найтроджен), расход энергии несколько выше, соответственно составу газа и способу очистки. Наибольшие перспективы на будущее имеет схема высокого давления порядка 800 atm с аммиачным охлаждением во вторичной конденсации с высоким рабочим напряжением в стенках кожуха и высокой скоростью газового потока. Подобная схема особенно целесообразна при высокой ценности специального металла и сравнительной дешевизне энергии в виде электрического тока. Низкие давления возникли в обход патента BASF (Габер-Бош), закреплявшего за этой фирмой все давления свыше 100 aim. Низкие давления в настоящее время никаких преимуществ перед высокими и средними с искусственным охлаждением не имеют. Сверхвысокие давления (5 000 atm) обещают 97% NH3за один проход катализатора, но они еще не вышли из лабораторной стадии.
Применение. А. играет колоссальную роль в качестве сырья для получения азотной кислоты и ряда азотсодержащих туков. Проблема А. является частным случаем решения проблемы азота. В настоящее время в промышленном масштабе связывание атмосферного азота и переработка его на вещества и удобрения происходят наиболее экономичным путем с помощью синтеза А. из элементов. Причина преимущественного развития синтеза А. из элементов коренится в след, сопоставлении: на 1 тонна связанного азота требуется энергии в виде тока или топлива в единицах У. Т.: 1) синтез А. из элементов 3—13 (в зависимости от метода производства азото-водородной смеси); 2) синтез азот ной кислоты в дуге 64; 3) синтез А. через цианамид 13;
4) синтез А. через нитрид алюминия 5.
Азотнокислотная промсть, являясь необходимым звеном в производстве средств обороны, в то же время служит источником получения азотистых удобрений. Эта особенность азотных установок позволяет балансировать производство удобрений и обслуживание военных нужд почти на неизменных капиталовложениях. Экономически неизбежным этапом современной азотной техники является синтез аммиака из элементов. Что касается вспомогательных и перерабатывающих цехов, то существует ряд возможностей производственного их комбинирования. Например при основном целевом назначении з-да— производстве туков—весь А. может идти в пере-работку на сульфат или хлорид аммония; при необходимости поставлять нитрующие средства требуется в составе завода наличие цеха окисления А. в окись азота, к-рый теперь также экономически неизбежен при переработке А. в азотную к-ту. 3-ды синтеза А. как правило строятся вблизи энергетич. базы. Источником азота является воздух (прямо или косвенно); источником же водорода в зависимости от характера энергетич. базы служат вода, уголь или нефтепродукты. Многообразие способов производства азото-водородной смеси, в значительной степени отражающее естественные условия установок, в кон-це-концов завершается процессом синтеза из элементов, все модификации которого в технологическом масштабе по существу весьма сходны.
Статистические данные по связанному азоту и А. Производство и потребление азотистых соединений в капиталистич. странах и уд.· в основных способов производства видны из табл. 5.
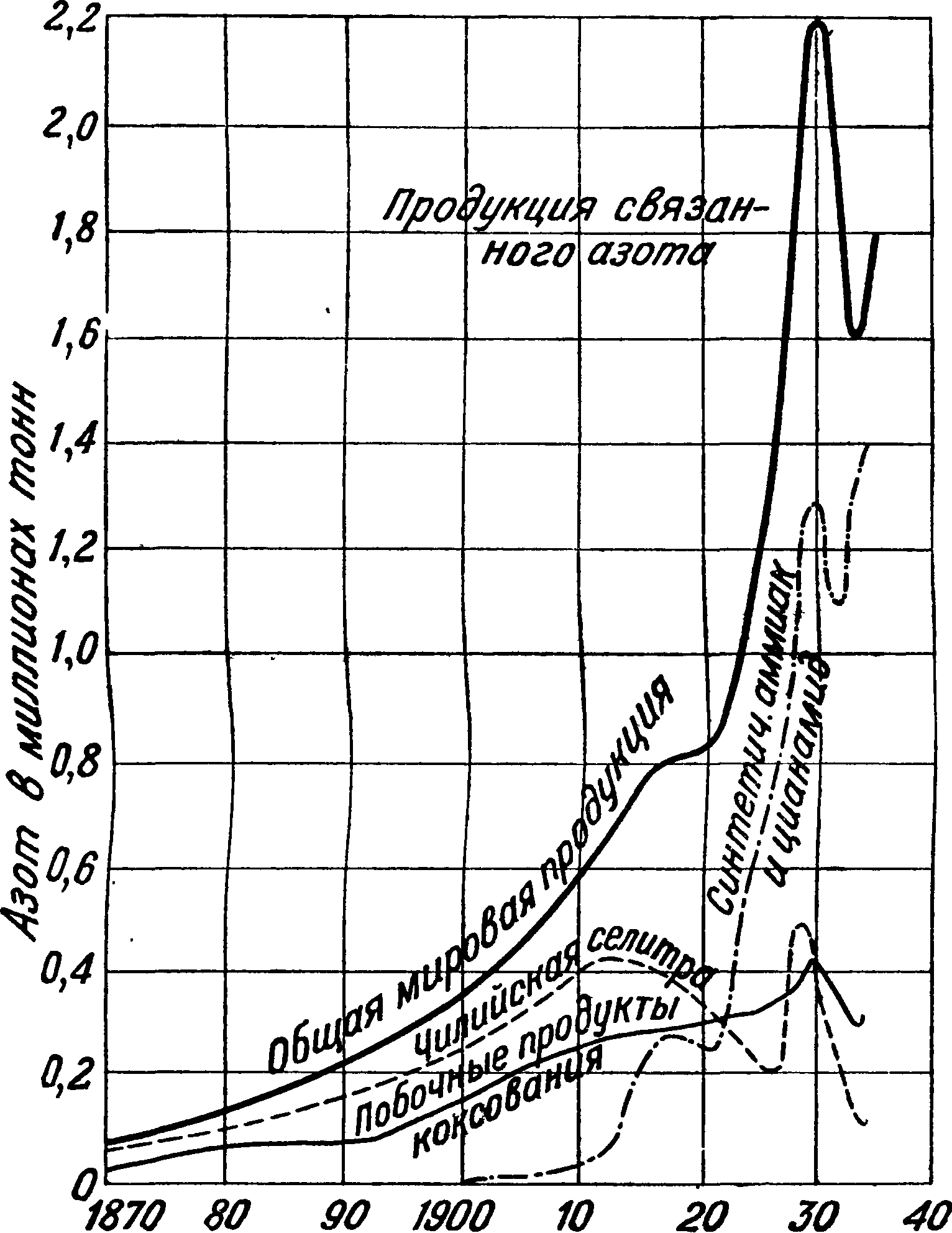
Рост мировой продукции связанного азота, начиная с 1870 г., показан на фигуре 8.
В СССР производство синте-тич. А. создано в течение первой пятилетки и получило дальнейшее мощное развитие в течение второй пятилетки. В 1932 г. производство син-тетич. А. базировалось на получении водорода путем конверсии водяного газа в размере 25% и по железопаровому методу в размере 75%. В 1937 г. уд. в производства по конверсионному методу составит 44,8%, по методу глубокого охлаждения— 35,6%, по парокислородному методу—7%, по электролитическому—8% и на долю устаревшего и дорого стоящего железспарового метода придется лишь 4,6% от всего количества производимого синтетич. А.
Необходимо отметить, что ряд народнохозяйственных проблем следует решать в комплексе с производством аммиака синтезом из элементов. Сюда относятся: полное использование ароматич. углеводородов коксового газа с· заменой цикла бензинё вымораживанием, получение серы при регенерации адсорбентов сероводорода, синтез метанола и высших ов на базе водяного газа и водорода (конверсионного или из другого
Таблица 5. — Производство и потребление азотистых соединений в капиталистических странах (в тыс. тонн). *
| Показатели | 1913 | 1928/29 | 1929/30 | 1930/31 | 1931/32 | 1932/33 | 1933/34 |
| I. Производство | |||||||
| Синтетич. аммиак.. | 7 | 485 | 442 | 349 | 522 | 560 | 540 |
| Цианамид кальция. | 38 | 192 | 264 | 201 | 135 | 168 | 192 |
| Норвежская селитра. | 15 | 136 | 131 | 111 | 79 | 118 | 106 |
| Прочие виды синтетич. азота. | — | 434 | 479 | 424 | 378 | 502 | 557 |
| Всего синтетич. азота. | 60 | 1 247 | 1 316 | 1 085 | 1 114 | 1 348 | 1 395 |
| Аммиак коксовых и газовых за | |||||||
| водов .. | 284 | 376 | 424 | 360 | 302 | 258 | 306 |
| Чилийская селитра. | 430 | 490 | 464 | 250 | 170 | 71 | 85 |
| Всего азотистых соединений
II. Уд. в основных способов производства (в%) |
774 | 2 ИЗ | 2 204 | 1 694 | 1 585 | 1 677 | 1 786 |
| Синтетич. азот..
Аммиак коксовых и газовых за |
70,7 | 59,0 | 59,7 | 63,1 | 70,2 | 80,4 | 78,1 |
| водов .. | 36,7 | 17,8 | 19,2 | 21,3 | 19,1 | 15,4 | 17,1 |
| Чилийская селитра.
III. Потребление |
55,6 | 23,2 | 21,1 | 14,8 | 10,7 | 4,2 | 4,8 |
| Синтетич. азот и аммиак коксов. | |||||||
| и газовых заводов. | — | 1 453 | 1 587 | 1 377 | 1 417 | 1 620 | 1 701 |
| Чилийская селитра. | — | 419 | 364 | 244 | 138 | 127 | 161 |
| Всего потреблено. | 1 _ | 1 872 | 1 1 951 | 1 621 | 1 555 | 1 747 | 1 862 |
| В том числе в сельскОхМ хозяйстве j
1 |
1 -
i |
1 684 | j1750 | 1 455 | 1 412 | 1 586 | 1 663 |
|
* Сельскохозяйственные годы (с 1/YIII по 31/VII); 1913 г. — календарный год. | |||||||
источника), получение нашатыря как побочного продукта производства соды и прочие Наоборот, аммиак может получаться как побочный продукт на базе колошниковых газов доменного процесса с кислородным дутьем и тому подобное. Долгое время метод синтеза А. через нитрид алюминия считался неудобным для осуществления, но сейчас вновь разрабатывается. Этот метод в основном является процессом получения окиси алюминия, необходимой для выработки металлич. алюминия. Сырьем для него служат кокс, боксит и азот.
Годы
Фигура 8.
Технич. трудности процесса—гл. обр. в конструкции печи, которая должна обеспечивать равномерную t° (ок. 1 600°). Перспективы этого способа м. б. уяснены из следующего сравнения: расход энергии в килограммах У. Т. на 1 тонна NH3 и на
1,6 тонн А1 (в виде 3 тонны А1203) составляет:
По нитридалюмиыиевому способу, считая в печи кпд 30%.. 5 000
При изготовлении А. и окиси алюминия порознь .. 23 000
а) аммиак синтезом (в среднем). 3 000
б) окись алюминия (по Кузнецову-
Жуковскому) .. 20 ООО
Основная реакция в печи:
Α1203+30+Ν2=2Α1Ν-{-30α -213 000 Cal.
Соотношения А. и алюминия взяты согласно реакции омыления нитрида
A1N+3H20=AH0H)3+NH3,
поскольку выход А. здесь практически количественный.
Цианамидный процесс хотя· и вырос за годы войны, но с тех пор не получил дальнейшего развития. Ни энергетический выход ни качество даваемого им А. не стимулируют в настоящее время его роста. Использование А. из газа коксовых печей в абсолютных цифрах увеличивается, поскольку этот процесс представляет собой один из элементов рационального баланса коксования. Новые коксовые установки в сопутствующих им химич. заводах обычно снабжаются скрубберами, улавливающими бензол и А., а равно и дистилляционными колоннами для укрепления аммиачной воды с 3 до 17%, если абсорбция NH3 идет не в кислой среде. Однако этот процесс потерял ведущую роль для снабжения народного хозяйства азотными продуктами внутреннего происхождения, хотя выход аммиака (1,2—1,5% от веса угля) и раньше не позволял рассматривать коксование иначе как способ, дающий аммиак в качестве побочного продукта.
Лит.: i)"Emmett, В г u η а и е г, «J. Am. Chem. Soc.», 1930, 7, р. 2682; 2) Bosch, «Ind. Chemist», 1934, p. 90; 3) в lumenthal, «Chimie et Industrie», 1934, 4, p. 972; *) Kalish, Ch. Markets, XII, 1931; 6) F а и s e r, «G-iorn. Ch. Ind. Appi.», 1931, 8; 6) D о d g e, «Ind. Eng. Ch.», 1932, 12, p. 1353. — Фокин Л., Синтез аммиака, Л., 1932; Голованов и Маляре»· с к и й, Синтез аммиака, Харьков, 1929; Ernst, Fixed Nitrogen, L., 1928; Curtis, Fixed Nitrogen, Ν.Ύ., 1933; W a e s e r, Die Luftstickstoffindustrie, Lpz., 1932; Tongue, The Design a. Construction of High-pressure Chemical Plant, L., 1934; W a e s e r, «Metall-borse», 1930, 9, p. 229; В о r e 1 1 i, «Ch. Met. Eng.», 1932, p. 126; Toniolo, Giammarco, «Шот. Ch. Ind. Appi.», 1933, p. 219; Gillespie, Beat-tie, «J. Am. Chem. Soc.», 1930, p. 4239; Scholvien, «Chem. Met. Eng.», 1931, p. 82, 133; С 1 a u d e, «Z. ang. Chemie», 1930, p. 417; «Chem. Met. Eng.», 1920, 8, p. 481; F i r m i n, «L’Industriechimique», 1924, v. 11, p. 200, 440; 1925, v. 12, p. 11, 208, 255, 446; 1926, v. 13, p, 8, 154, 251;.OCT 3753 и 3754. Ю. Севастьянов.