 > Техника, страница 13 > Анилин
> Техника, страница 13 > Анилин
Анилин
Анилин, фениламин, аминобен-з о л — простейший амин ароматического ряда, строения CeHsNHa. Впервые А. был получен при сухой перегонке индиго в 1826 г., а в 1834 г. был найден Рунге в каменноугольной смоле и назван им «кианоль» благодаря свойству давать синее окрашивание с раствором белильной извести. В том лее году Митчерлих нитрацией бензола получил нитробензол, из которого Η. Н. Зинин в 1842 г. в Казани при действии сернистого аммония получил А., которому дал название «бензидам». Истинное строение А. было установлено в 1843 г. А. В. Гофманом. Открытие Зинина и работы Гофмана позволили от лабораторного приготовления анилина сухой перегонкой индиго перейти к заводскому производству из бензола через нитробензол.
А.—маслянистая жидкость, бесцветная в чистом состоянии, но быстро буреющая при наличии незначительных примесей при стоянии; уд. вес при 15° 1,0268, t°Kun.
184,3 — 184°,4, t°3acm.—6°,24, показатель преломления 1,585 при 20°. А. смешивается во всех отношениях с голем, эфиром, бензолом и тому подобное. 100 ч. воды растворяют при 15° 3,611 ч. А. При этой же t° 100 ч. А. растворяют 5,12 ч. воды. С повышением t° взаимная растворимость А. и воды растет и достигает критической точки при 168°. А. значительно растворим в растворе солянокислого А.; перегоняется с водяными парами, и паровая фаза имеет t° 99—99°,5 при содержании 20—21% А. и 79—80% воды. Более подробные сведения о физич. свойствах А. см. [*] и [8]. По своим хим. свойствам А.— органическое основание и дает с к-тами соли, из которых наиболее важ ной является солянокислый А.—анилиновая соль. А. является весьма реакциеспособным веществом. При действии на А., даже в разбавленных растворах, белильной извести получается интенсивное синее окрашивание. При действии двухромовокалиевой соли на холоду получается хинон (смотрите). При действии азотистой кислоты или ее солей в кислой среде получается диазосоединение (смотрите). Водородные атомы в молекуле А. могут замещаться различными остатками, причем в реакцию могут вступать как водородные атомы бензольного ядра, так иво-дородные атомы амидной группы. Наиболее важными реакциями является введение в ядро остатков сульфо-группы, галоидов и нитро-группы. Замещение водородн. атомов амидной группой может наступить за счет кислых (ацильных), жирных (алкильных) и ароматических (арильных) остатков и ведет к получению весьма важных продуктов, как: ацетанилид, диметиланилин и диэтил-анилин, дифениламин и др. Более подробно о хим. свойствах А. и его производных см. С3], [4] и [«]. А. и его производные весьма важны в техническом отношении, т. к. служат исходными продуктами для получения красящих веществ (смотрите), фармацевтических продуктов, как, например, сальварсан и др., чатых веществ, как тетрил и тому подобное. О техническом применении А. см. С6] и П- А. в весьма широких размерах находит применение на текстильных фабриках для непосредственного получения прочных черных окрасок на хлопчатобумажной ткани (смотрите Черный анилин). Продажный А. бывает различных технич. качеств и в зависимости от примесей имеет следующие продажные термины: А. для синего содержит 99—99,9% чистого А., кипит в пределах 182—184°, служит для получения синих красителей, как, например, о-вый синий, щелочной голубой, индиго (смотрите) и др.; А. для красного содержит почти эквимолекулярное соотношение А., о-то-луидина и и-толуидина, кипит в пределах 190 —198°, служит для получения красных красителей типа фуксина (смотрите); А. для сафранина содержит 35 — 50% А. и 65 — 50% о-толуидина и служит для получения сафранина (смотрите). Обыкновенно А. для сафранина является отгоном при получении фуксина и имеет название «Fuchsin ёейаррб». Впрочем, в настоящее время на рынок поступает в большинстве случаев только А. для синего, из которого готовят на самих красочных заводах другие сорта простым смешением его с соответствующими количествами о- и w-толуидинов, тоже имеющихся в продаже в чистом виде.
Производство А. основано на опыте Зинина (смотрите выше), но в качестве восстановителя употребляется не сернистый аммоний, а более дешевый материал—железные или чугунные стружки в присутствии уксусной или соляной к-ты. Было замечено, что реакция восстановления нитробензола железом идет вполне гладко при наличии г/60 от потребного теоретически количества кислоты. Объяснение этого найдено в каталитическом действии FeCl2, благодаря к-рому взятое в качестве восстановителя железо превращается в Fe304, а нитробензол восстанавливается в А. Еще О. Витт в 1887 г. предложил следующее подтверждающееся и в наше время химическое объяснение:
4С„ HsN02 + 4Н20 + 24FeClo
4C6N5NH2 + 12Fe2Cl40;
9 Fe + 12 Fe2Cl40 24 FeCl2 + 3 Fe304. Нет необходимости вводить в реакционную смесь свободную соляную кислоту, а можно ее заменить соответствующим количеством раствора солянокислого А. или готового FeCl2. Реакция восстановления нитробензола идет с сильным выделением тепла и выразится суммарно следующим ур-ием:
4CeH6NO, +4Н20 + 9Fe->
4- 4C6HSNH2 + 3Fe804 -f - 563Cal.
На з-дах, производящих А., восстановление нитробензола ведется в соответствующих котлах-редукторах вертикального или горизонтального типа, снабженных мощной мешй, арматурой для пропускания острого пара, воды и для загрузки железных стружек. Мешалка редуктора должна приводить в движение всю массу железных стружек в смеси с Fe304, к концу реакции лежащих на дне редуктора, чтобы не дать им слеживаться. Редуктор соединен с обратным холодильником в виду того, что реакция восстановления идет при кипении всей смеси. Редукторы снабжены спускным краном для удаления Fe304 в смеси с непрореагировавшим железом и для промывки аппаратов. Обычным материалом для редукторов служит чугун, причем иногда он снабжается внутренней кислотоупорной футеровкой, что увеличивает продолжительность службы аппарата. Процесс получения А. может быть разбит на 3 стадии: 1) восстановление нитробензола, 2) извлечение А. из редукционной смеси и 3) очистка А. Восстановление нитробензола в заводском масштабе ведется следующим образом. В редуктор загружается нитробензол, часть железа и вся соляная к-та. Туда же заливается вода, и смесь подогревается острым паром, при движущейся мешалке, до кипения, после чего пуск пара, вследствие экзотермичности реакции восстановления, прекращается. t Интенсивность кипения смеси регулируется притоком нитробензола и загрузкой железа. Идеальными условиями ведения процесса является равномерная загрузка нитробензола и железа в течение всего процесса восстановления; но в техническом выполнении это представляет определенные трудности, и поэтому обыкновенно весь нитробензол заливают в редуктор при начале процесса, а железо загружают небольшими порциями, от времени до времени, через загрузочную воронку специального устройства. Для восстановления берется обычно до 30% избытка железа. Кипящая смесь А. с водой и нитробензолом, охлаждаясь в обратном холодильнике, стекает в редуктор; по количеству и цвету дистиллата наблюдают за ходом реакции. Т. к. ничтожные следы нитробензола окрашивают дистиллят в желтый цвет, появление бесцветного дистил лата служит признаком конца реакции. Обычно последние остатки нитребонзола медленно восстанавливаются, и для доведения реакции до конца необходимо поддержать кипение смеси дополнительным пропусканием острого пара. В зависимости от качества взятых продуктов и от количества восстанавливаемого нитробензола, время реакции бывает различно. В редукторах обычных размеров для восстановления 1 000—1 500 килограмм нитробензола процесс восстановления длится 9—12 ч. Примесь бензола в нитробензоле значительно замедляет скорость восстановления. На скорость процесса влияет также и степень измельчения и свойства взятого железа. Опилки вступают в реакцию быстрее стружек, но т. к. в опилках возможно присутствие землистых и иных примесей, предпочитают им мелкие стружки. Чугунные стружки являются более реакциесгюсобиыми, чем железные; наименее пригодными для восстановления являются стальные стружки и опилки. Когда процесс восстановления окончен, в редуктор загружается щелочь для нейтрализации взятой соляной к-ты, цри чем наиболее употребительной является известь. После этого в редукторе находится смесь из А., воды, хлористого кальция, непрореагировавших железных стружек и Fe304, и начинается вторая стадия процесса—извлечение А. из редукционной смеси. Извлечение А. из редукционной смеси м. б. достигнуто след, методами: 1) перегонкой А. из редуктора с водяными парами, 2) отфильтровыванием А. вместе с водой от железных стружек и Fe3Q4и 3) высаливанием и сифонированием А. Самым старым, но еще не вполне устарелым, является метод перегонки А. с водяными парами. Для этого, немедленно после нейтрализации содержимого редуктора, последнее приводится пропусканием острого пара в кипение. Пары А. и воды, проходя через холодильник, конденсируются и поступают в приемник. Для извлечения 1 000 килограмм из редуктора требуется затрата около 6 000 килограмм пара с соответствующим количеством воды для охлаждения холодильников. В редукторе остается ок. 0,5% А. Собранные в приемник 7 000 килограмм жидкости, содержащей 1 000 килограмм А., при охлаждении расслаиваются на два слоя, из которых в верхнем находится раствор 40—50 килограмм воды в 800 килограмм А., в нижнем же 6 000 килограмм воды и около 200 килограмм А. В делительной воронке анилиновый слой отделяется и поступает на очистку, водный же слой поступает на регенерацию из него А. Метод фильтрования является более дешевым, т. к. не требует большого расхода пара для перегонки и воды для охлаждения, но существенным его недостатком является трудность фильтрования и необходимость значит, промывки и продувки на фильтр-прессах полученных осадков железа и Fe304. И при этом методе А. в приемнике получается в присутствии значительных колич. воды, растворяющей 10—20% полученного А., и потому нуждается в регенерации. Метод сифонирования состоит в высаливании А. в редукторе поварен, солью и в отделении сифонироваиием всплывающего наверх А. Для этого в редуктор засыпают поваренную соль для получения 10—15% раствора ее. При t° 80 — 90° большая часть А. всплывает наверх, а в нижнем водном слое, над отстоявшимся железом и Fe304остается при этих условиях 0,3—0,5% А; вакуум-сифоиом верхний анилиновый слой передается в приемник, откуда поступает на очистку. Недостатком этого метода яв-
перемешивается с равным по весу количеством нитробензола, и отстоявшийся нижний слой нитробензола с извлеченным анилином вновь поступает в редуктор для восстановления. В верхнем водном слое остаются в растворе 0,25% нитробензола и 0,35% А. Получившаяся во всех случаях вода всегда содержит нек-рое количество А., и, для возможно более полного использования его, служит для питания редукторов и паровых котлов, снабжающих редукторы острым паром. В зависимости от местных экономических условий, для достижения наиболее полного и наиболее дешевого получения А. приходится сочетать один из методов извлечения А. из редуктора с одним из методов регенерации А. из анилиновой воды. В качестве иллюстрации приведем таблицу хода процесса в редукторе при сочетании метода сифонирования с дополнительной отгонкой оставшегося в редукторе А. водяными парами по [3]. Загружено 1 125 килограмм нитробензола, 180 килограмм железных стружек, 80 килограмм технической соляной кислоты и 800 килограмм воды. В течение процесса восстановления было дополнительно загружено небольшими порциями еще 1 170 килограмм железных стружек.
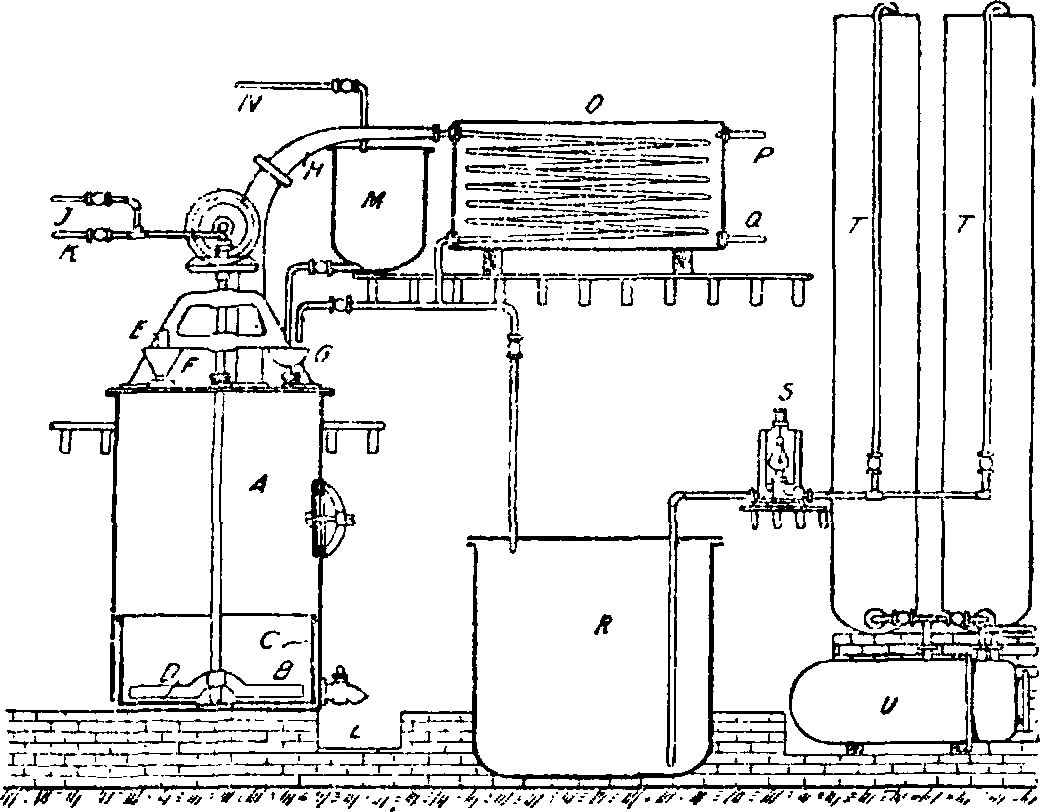
Установка анилинового производства: А—котел редуктор, В—мешалка, С—чугунная обкладка стен, I)—чугунное днище, Е—деревянная пробка,
Е—наиолшггельная воронка, G—подвод нитробензола, Н—трубопровод к холодильнику, J—трубопровод для чистого пара, Д—трубопровод для наров, содержащих анилин, L—кран для выпуска железного шлама, М —сосуд с нитробензолом, N-трубопровод, подводящий нитробензол, О—холодильник, Р—отвод нагретой воды, Q—впуск холодной воды, К—приемник, 5—паровой насос,
Т—колонки для разгонки анилина, U—прибор для нагнетания анилинсодержащей воды в паровой котел. ляется неполное отстаивание А., в силу чего, для возможно более полного извлечения А., приходится оставшийся после сифонирования в редукторе А. извлекать одним из вышеуказанных приемов, лучше всего перегонкой с водяными парами. Регенерация А. из водного его раствора, носящего название анилиновой в о-д ы, играет весьма существенную роль, т. к. в нем содержится 3—4% по весу А., или 10—20% всего полученного А. Для извлечения А. прибегают к следующим приемам: 1) разгонке анилиновой воды в колонках, 2) высаливанию А. из анилиновой воды и 3) извлечению А. из водного раствора органич. растворителем, в частности—нитробензолом. Разгонка анилиновой воды становится возможной в силу того, что паровая фаза смеси А. с водой содержит 20—21% А., в то время как жидкая фаза содержит 3—4% А. и производится на мощных колонках тарелочного или насадочного типа. Для извлечения 100 килограмм А. из 3 000 килограмм анилиновой воды требуется, в зависимости от системы колонки, от 5 000 до 6 000 килограмм пара, что тяжело сказывается на калькуляции А. Более дешевым является высаливание А. поваренной солью из анилиновой воды, в растворе которой после высаливания остается 0,3—0,5% А. Изящным является метод извлечения А. из воды нитробензолом. По [8] анилиновая вода
Загрузка
Подогрев острым паром до кипения.
Восстановление ..
Подогрев острым паром для конца реакции
Нейтрализация ..
Высаливание 25 0 килограмм поваренной соли.
Отстаивание и сифонирование.
Отгонка оставшегося А. 2 000 килограмм пара. Разгрузка и промывка редуктора.
| а/« | час |
| V. | » |
| 51/, | |
| IV. | |
| V, | » |
| V. | » |
| V* | » |
| 4 | )> |
| 7» | » |
Всего.14 часов.
Полученный по любому методу А. содержит в растворе до 5% воды и нуждается для получения чистого продукта в очистке. Последняя достигается вакуум-перегонкой из перегонных кубов. Перегоняющаяся вначале вода с А. поступает на регенерацию по любому из вышеуказанных способов; технически же чистый 99—99,9%-ный А. собирается отдельно в приемном резервуаре, откуда после исследования разливается в железные бочки и идет для продажи или для производства любых производных А. Восстановление нитробензола железными стружками является громоздким и сложным процессом и заставило искать иных путей получения А. Нитробензол может быть восстановлен электролитически в А. по [®], [10], [“] и [1а] в электролизерах с диафрагмой в гольно-водной среде в присутствии соляной и 30%-ной серной к-ты, при оловянных, медных, платиновых катодах или при катодах из индифферентного материала, при наличии в ванне солей олова, меди или медного порошка. Каталитическое восстановление нитробензола в А. водородом основано на работах Сабатье и Сандерена [“], показавших, что нитробензол и водород при пропуске над активированным никелем при 250° дают А. Дальнейшее повышение t° ведет к более глубокому расщеплению—до бензола, аммиака и даже метана. По [“] через нитробензол, нагретый до 120°, пропускается водяной пар, увлекающий с собой пары нитробензола. Их смесь вместе с водородом проходит через нагретую до 120° трубку с никелем с такой скоростью, чтобы из другого конца трубки уходили пары А. и воды без примеси нитробензола. Никелевый катализатор м. б. заменен платиной или палладием. Существенным недостатком этого приема является получение А. в смеси е значительным количеством воды, что вызывает необходимость регенерации его из раствора и тем самым увеличивает стоимость его. По [16], [16], Р7] и [18] пары нитробензола в смеси с избытком водорода или водяного газа пропускаются через нагретые до 200—220° трубки с катализатором при скорости, соответствующей выходу из другого конца трубки паров А. без примеси нитробензола, чем достигается теоретический выход. Пары А. конденсируются, а избыточный водород вновь идет на восстановление. В качестве катализатора м. б. взяты специально приготовленные смеси меди и цинка, меди и железа, меди и магния, меди и серебра, а также и кислородные соединения железа. Этот метод каталитического восстановления дает возможность осуществить непрерывный ход процесса и делает ненужной регенерацию А. Техническ. применение этого метода зависит от получения недорогой каталитически стойкой массы катализатора. Иные методы каталитического восстановления по [19] и [20] не представляют существенного интереса, т. к. ведутся при давлении от 15 до 200 atm и требуют периодической загрузки и разгрузки автоклавов. Не лишен интереса путь восстановления нитробензола двусернистым натрием Na2S2, т. к. при пользовании им в качестве отброса процесса получается легко кристаллизующаяся серноватистонатриевая соль. При наличии дешевого хлоробензола, А. м. б. получен из последнего по [22] действием водного раствора аммиака в автоклаве при 180° в течение 24 ч. Производство А. по любому из методов требует сугубой осторояшости в силу ядовитых свойств А. При вдыхании паров последнего, даже в слабых концентрациях, наступают симптомы отравления, заключающиеся в посинении губ и слизистых оболочек, головокружении, потере аппетита, чувстве усталости и слабости. В более серьезных случаях наступает явление затрудненного дыхания, похолодение всего тела и обморочное состояние. Непрерывное пребывание в течение долгого времени в атмосфере, содержащей анилиновые пары, ведет к частым головокружениям, анемии и острой неврастении. В виду этого необходимы: герметичность аппаратов, хорошо действующая вентиляция, снабжение рабочих специальной одеждой, достаточное количество умывальников и подробное инструктирование рабочих о мерах предосторожности. В СССР А. производится на з-дах Анил-треста в Кинешме и Дорогомиловском в Москве; на заводе красочного треста «Красный химик» в Ленинграде производство А. ведется по методу восстановления нитробензола железными стружками. По [23] потребность России в А. и его солях до войны выражалась в 166 000 п., из которых 61 000 п. ввозилась из-за границы. В настоящее время вся потребность м. б. удовлетворена производством внутри страны. Цена А. в довоенное время колебалась от 10 до 12 р. за и.; наиболее дешевым источником А. была Германия, где в 1912 году А. стоил 1,2 мар. кг, или ниже 9 р. п. Цены на А. в настоящее время приближаются к довоенным. В Америке А. стоит 15 центов англ, фунт, или около 70 коп. кг.
Лит.: ) «Lieb. Ann.», В. 447, § 31, Lpz., 1926; 2)Beilstein F., Handbuch d. organischen Chemie, 3 Aufl., B. 2, p. 308, Hamburg, 1896; s) Grog-gins P. H., Aniline and its Derivatives, L., 1924; ) Mey er Y. u. J akobson P„ Lehrbuch d. organ. Chemie, B. 2, T. I, p. 164, Lpz., 1902; s) Ворожцов Η. H., Ступени в синтезе красителей (Химия циклич. промешут. продуктов), Л., 1926;*) В urn-Ьагу Η. М. a. Davidson A., The Industrial Applications of Coal Tar Products, L., 1925; 7) Schotz
S. P., Synthetic Organic Compounds, p. 212, L., 1924; 8) Г. П. 282 531; ») Г. II. 118 942; 10) Г. П. 117 007; ll) Г. П. 130 742;13) Г. II. 131 404; is) «CR», 135, p. 226: “) Г. П. 282 492; i5) Г. II. 283 449; “) Г. II. 282 568; 17) Г. П. 273 322;18) Г. П. 331 303; 1B) Г. П. 281 110; 20) Г. П. 486 064; 21) Г. ΓΙ. 144 809; 22) Г. П. 204 951; 23) Фокин Л. Ф., Обзор хим. промышл. в России, Научн. хим.-техн. изд., 1922. И. Иоффе.