 > Техника, страница 51 > Катализ
> Техника, страница 51 > Катализ
Катализ
Катализ, изменение (в обычном понимании—увеличение) скорости химич. реакций в присутствии посторонних веществ, т.н. катали за торов (Ктр.), остающихся после завершения реакции в массе (макроскопически) неизмененными. К.—чрезвычайно распространенное явление. Известно очень мало реакций, ход которых не зависит от присутствия посторонних веществ. Соли, к-ты, щелочи, порошкообразные металлы, окислы, стенки сосуда, влага, среда, в которой идет реакция,—все это оказывает влияние на скорость последней, так что практически «чистые» случаи химической кинетики (смотрите Кинетика химическая) встречаются как исключение. Часто термином К. объединяют все явления возрастания скорости реакции, каковы бы ни были причины этого ускорения. Вследствие множественности причин, вызывающих явления К., нет общей теории К., которая была бы применима ко всем реакциям: механизм, пригодный для объяснения одной реакции, часто неприменим для дру-
гой. Обычно сводят действие Ктр. к образованию промежуточных легко распадающихся веществ, или же функцию Ктр. усматривают в том, что благодаря его присутствию реагирующие вещества переходят в нек-рое особое состояние, облегчающее ход реакции. Современное состояние вопроса о К. таково, что в нек-рых случаях б. или м. известен механизм реакции, но это знание скорее индуктивное, и на основании его нельзя количественно предвидеть ускоряющее действие того или иного вещества на реакцию. Явления К. имеют большое значение в химии, т. к. они 1) охватывают значительный круг реакций, 2) необходимы для уяснения природы химич. взаимодействия и 3) имеют большое применение в технике, где зачастую осуществление процесса в промышленном масштабе сводится к подысканию подходящего катализатора.
К. п термодинамика. Основным вопросом К. является следующий: можно ли при любых заданных условиях (t°, давление и концентрации исходных веществ) найти Ктр., к-рый ускорил бы реакцию в желательном направлении, прямом или обратном. Согласно 2 принципу термодинамики, химич. реакции идут в сторону уменьшения свободной энергии системы (или в сторону возрастания энтропии) до тех пор, пока не будет достигнуто нек-рое конечное состояние равновесия между исходными и результирующими веществами. С момента достижения этого состояния концентрации перестают измен яться, и тогда система веществ находится в динамическом стационарном (в смысле концентраций) равновесии (смотрите Действующих масс закон), характеризуемом для каждой t° константой равновесия, то есть определенным соотношением концентраций всех входящих в реакцию веществ. При химич. ур-ии типа пА + тВ А иС + rD
в простейшем случае, когда вещества подчиняются законам идеальных газов, имеем:
[А]п [В]™ к
(1)
где К—константа равновесия, а величины в скобках — концентрации соответственных веществ при равновесии. Значение К как ψ-ии ί° определяется термодипамич. ур-ием изохоры реакции
din К Q cLT ~ НТ ’
(2)
где Т — абс. темп-pa, Q — тепловой эффект реакции, R—газовая константа, отнесенная, как и Q, к 8-молю и равная 1,98 cal/г. При Q= Const (что верно лишь в небольшом Р-ном интервале) ур-ие (2) дает:
Q
К=Ре
RT
9
(3)
где Р—константа, введенная интегрированием. Течение реакции направлено в сторону достижения таких концентраций, при которых выполняется ур-ие (1). Теория К. должна считаться с постоянством константы К, т. к. возможность нескольких значений К при одной ί° противоречит 2-му принципу, являясь случаем perpetuum mobile 2-го рода. Химическая кинетика показывает, что К
есть отношение констант скоростей обратной (/с2) и прямой (7сг) реакций: К=^ Если скорости этих взаимно противоположных реакций обозначить через v1 и г>2, то
1 (4)
v,-k2CucC£
где обозначения С с индексами показывают соответствующие концентрации. С течением времени последние принимают такие значения, что, при данных 7«х и /с2, vx становится равным г>2. Это приводит к выражению:
[А}« гву» _ fu _ „
[С]« IDJ»· k, Λ· W
Функция Ктр. заключается в том, что в его присутствии реакция скорее доходит до состояния равновесия. Термодинамика дает возможность найти приближенное значение К из теплового эффекта, теплоемкостей и химич. констант. Если расчет (например по приближенному ур-ию Нернста) показывает, что намеченная реакция связана с уменьшением свободной^энергии, то имеет смысл искать какого-нибудь Ктр. для ускорения реакции. Возможность кажущегося изменения константы равновесия от количества внесенного Ктр. (например при гидролизе этилацетата в присутствии соляной к-ты) объясняется тем, что Ктр. влияет на коэфф-ты активности (смотрите Диссоциация электролитическая) составных частей, так что реальные концентрации (активные массы) в присутствии Ктр. и в его отсутствии могут быть различными. Реакция протекает и без Ктр., приводя к тому же количественному соотношению, но на это требуется больше времени. При наличии веществ в концентрациях, находящихся в равновесии, Ктр. не может оказывать какого-либо влияния. Увеличивая константу скорости прямой реакции кг, Ктр. должен увеличить соответственно и 7«2, что вытекает из неизменности отношения 7с2: кг. Один и тот же Ктр. может ускорять либо прямую либо обратную реакцию, в зависимости от того, с какой стороны реакция подходит к равновесию. Платина или палладий, употребляемые при гидрогенизации бензола в циклогексан, применяются при ί° выше 200° для ускорения противоположной реакции, то есть для превращения циклогексана в бензол; первичные и вторичные ы, получаемые путем гидрогенизации альдегидов и кетонов при 150—180° с медью или никелем в качестве Ктр., регенерируются при ί° 250—300° в присутствии того же Ктр.
К. и химическая кинетика. Теория химич. кинетики приводит к следующему выражению для константы скорости:
_ Q,
кг=Рхе RT
_ Qa
7с2=Р2 е НТ Сопоставляя ур-ия (6) с ур-ием (3), находим:
Q — Q-г —Qit Р — · (О
В константу скорости входят факторы: ки-ыетич. (Pj, Р2) и энергетический (Qi, Q2)-Поскольку реакция осуществляется при столкновениях молекул, ее скорость должна за

висеть от числа последних, от расстояний, на которые должны подойти друг к другу молекулы для наступления реакции, от средней скорости движения молекул и от характера их соударения (двойное, тройное и т. д. столкновение). Все эти факторы находят отражение в члене Р1 и соответственно Р2. Факторы Q1 и Q 2 выражают энергию активации прямой и обратной реакций. Опыт показывает, что в каждый данный момент только незначительная часть из общего числа молекул способна к реакции (например, для случая распада пятиокиси азота, N205, только одна молекула из 1017 является активной), причем число таких активных молекул быстро возрастает с ί°. В связи с этим считают, что молекула должна в процессе реакции пройти через нек-рое промежуточное состояние, получая при этом избыточную энергию активации, которая уменьшает ее сопротивляемость, облегчая внутримолекулярную перегруппировку или распад. Длительность этого промежуточного состояния—порядка 10_s ск. Согласно современным представлениям о строении материи, атомы и молекулы могут существовать в ряде стационарных (квантовых) состояний разной степени устойчивости и разной вероятности взаимных переходов. В случае атомов—наиболее устойчивым (соответственно минимуму запаса потенциальной энергии) является такое состояние, при к-ром электронные орбиты наиболее близки к ядру атома. Сообщая атомам энергию, отвечающую т. н. резонансным потенциалам, можно заставить электроны перейти на более далекие орбиты. Аналогичные явления (в усложненной форме) имеют место, как показывают спектральные исследования, и в молекулах. Активация молекулы, то есть вызванный поглощением энергии переход электронов на более высокий квантовый уровень, влечет за собой особое состояние мета стабильности, когда прочная, нормальная связь составных частей молекулы ослаблена. Это состояние (п р е д-диссоциация) могло быть изучено фотохимически для ряда молекул благодаря тому, что активация, или возбуждение, м. б. вызвана поглощением света определенной длины волны. Причиной активации может явиться столкновение молекул, движущихся с большими скоростями. При этом м. б. учтена кинетич. энергия не только прямолинейного движения, но и вращательного и колебательного. Механизм реакции с активацией мыслится по схеме:
(8)
Нормальная молекула п в результате столкновения или другим путем получает энергию активации и переходит в возбужден-ное, активное состояние а. У образовавшейся активной молекулы есть две возможности:
1) она может столкнуться с молекулой исходного вещества п: тогда энергия активации рассеивается, распределяясь между двумя молекулами, и происходит д е з актив а ц и я (процесс, обратный активации);
2) она может самопроизвольно распасться на два активированных продукта реакции о,и а"; последние, сталкиваясь с нормальными исходными молекулами, отдают им свою избыточную энергию и сами превращаются в нормальные молекулы конечных продуктов; за счет отданной энергии исходные молекулы активируются, и реакция продолжается прежним путем; такой механизм называется цепным механизмом и, возможно, имеет место в большинстве каталитич. процессов. В нек-рых случаях звеньями реакционной цепи м. б. атомы, образующиеся при активации. Причина активации может также заключаться в поглощении света (радиационная теория).
Увеличение константы скорости при наличии Ктр. может произойти либо при возрастании Рг либо при уменьшении Qx (ур-ие 6). Рг может увеличиться в том случае, когда реакция высокого порядка, то есть требующая одновременного столкновения нескольких молекул, может осуществляться в присутствии Ктр. через промежуточн. реакцию низшего порядка. При нормальных условиях число тройных столкновений примерно в 1 000 раз меньше числа двойных; если реакция 3-го порядка протекает с помощью Ктр. по ур-шо 2-го порядка, то при прочих равных условиях ее скорость будет значительно больше. Рх может также возрастать, если увеличивается радиус столкновения. В случае обыкновенных электронейтральных молекул взаимодействие их обнаруживается лишь при сближении на расстояние порядка молекулярных размеров (-10”8 см). В случае же ионов — силы притяжения растут и сказываются на больших расстояниях; вследствие этого на беспорядочное движение молекул налагается новое условие, и число столкновений возрастает. Фактор может уменьшиться, например, в том случае, когда промежуточная реакция протекает при меньшей энергии активации. Чем меньше избыток энергии, требуемый для приведения молекулы в активное состояние, тем больше число молекул, могущих активироваться при столкновении. Если распад молекулы АВ по ур-шо AB -s А + В требует энергии активации Qlr если, далее, АВ легко и быстро дает с Ктр. С комплекс АВС, распад которого АВС-+А + В + + С идет с энергией активации Q^-a, то при одинаковых условиях комплекс АВС
а распадается быстрее в евг раз. При средних значениях Т=500° и величине a ss 20 000 cal на з-моль (во многих опытах имеют место именно такие условия для ряда реакций на поверхности Ктр.) скорость возрастает в десятки млн. раз. Уменьшение Q1 может происходить при адсорбции, когда молекулы, налетая на поверхность адсорбента, становятся реа.кциеспо-собными. В табл. 1 приведен ряд случаев, когда реакция идет в присутствии Ктр. и без него; в первых случаях энергия активации значительно ниже.
Различают К. гомогенны й—когда Ктр. находится в одной фазе с реагирующими веществами, и гетерогенный. Последний приобрел большое техиич. значение. В этом случае реакция чаше всего происходит на поверхности твердого Ктр. Обычно Ктр. действует в незначительн. количестве: 1з-атом платинового золя оказывает ката-
Таблица 1.—Энергия активации пек-рых простейших реакций (Qt — в cal на г- моль).
| Реакция | Qj без Ктр. | QtB присутствии Ктр. | Катали затор |
| Распад NaO. | 58 500 | 32 500 1 | Платина
Золото |
| » Н.Т. | 44 000 | 25 000 | Золото |
| » NH„ | 70 000—80 000 | 39 000 | Вольфрам |
| Образование | |||
| H.S. | 51 400 | 25 750 | Кислород |
литич. действие на 7-107 л перекиси водорода; присутствие 10“13 г нормального раствора CuS04 ускоряет окисление сульфита натрия в сульфат; часто роль Ктр. играют малейшие следы воды и других веществ.
В качестве Ктр. в лабораторной и заводской практике употребляются восстановленные металлы (гл. обр. магний, алюминий, марганец, никель, железо, медь, серебро, золото, платина, палладий, осмий), металлоиды (сера, фосфор, хлор, бром, иод), окислы, минеральные к-ты, щелочи и многие другие неорганич. и органич. соединения. Способ приготовления Ктр. влияет на их эффективность; существ, значение здесь имеет характер поверхности, способ получения (мокрым или сухим путем, из тех или иных соединений данного элемента), t° обработки, сушки, прокаливания, наличие примесей. При гетерогенном К. обычно наносят Ктр. на подходящую основу, или подкладку, выбирая для этого вещество с сильно развитой поверхностью (кизельгур, активированный уголь, пемза, силикагель и др.); в частности, подкладкой может служить вещество, из которого готовится Ктр. При гидрогенизации над никелем получают хорошие результаты, когда исходная окись его восстановлена не нацело: т. о., здесь Ктр. является никель, распределенный на зернах его окиси. В целях получения Ктр. в наиболее раздробленном состоянии пользуются их коллоидными растворами (золями); вследствие малой устойчивости последних употребляют обычно золи, защищенные обратимыми (лиофильными) коллоидами. Золи Pt и Pd, защищенные ли-зальбиново- или протальбиновокислым натрием, нашли большое практич. применение (Ктр. Пааля, см. Гидрирование). Связанное с защитой обволакивание частичек Ктр. увеличивает и длительность его действия, понижая, однако, интенсивность последнего. При гомогенном К. известен ряд случаев, когда скорость реакции пропорциональна концентрации Ктр. (наир., скорость разложения диазосоединений в водном растворе пропорциональна концентрации водородных ионов, являющихся Ктр.); однако, как общее правило, эта зависимость более сложна. В гетерогенном катализе главными факторами являются строение,характер и специфичность поверхности. Физико-механическ. условия—скорость пропускания вещества над Ктр., перемешивание, способ наложения Ктр. на подкладку, размещение Ктр. в реакционном пространстве, скорость диффузии—имеют большое значение. Платина в виде черни, пластинок, тампона, сеткидействует по-разному, причем форма, пригодная для одной реакции (например для окисления сернистого газа), не пригодна для другой (например окисления аммиака). Эти факторы затрудняют точную воспроизводимость опытов. Макроскопически Ктр. после реакции остается таким же, каким был до начала; наблюдаются изменения лишь поверхностной структуры, например появление налетов ката-литич. пыли. Общей характеристикой Ктр. является то, что его количество не находится в стехиометрии, связи с количеством прореагировавшего вещества. Это не исключает возможности промежуточных образований, в особенности если принять во внимание цепной механизм К. или быстрое активирование на поверхности. При выборе Ктр. на практике исходят не только из учета его ускоряющей способности (одна и та нее реакция м. б. ускорена разными Ктр.), но также принимают во внимание дешевизну, легкость получения, чувствительность и капризность Ктр. к изменению условий. При синтезе аммиака уран, например, дает хорошие выходы, но обладает недостатком — чувствительностью к влаге, понижающей его действие; поэтому практически удобнее дешевое и более постоянно действующее железо, несмотря на меньший выход продуктов, получаемых при работе с этим Ктр. В начале действия Ктр. выходы растут, постепенно достигая максимума, но с течением времени изменение структуры, пористости, деформация поверхности от температурных влияний, загрязнения — влекут за собой понижение эффективности катализа.
Действие поверхности при К. В случае гетерогенного К. существенны явления, имеющие место при столкновении молекул с Ктр. Принято считать, что при этом происходят изменения физико-химич. порядка, в результате которых молекулы становятся более" реакциеспособными. Действие Ктр. связано с адсорбцией (смотрите) и м. б. весьма многообразным. Изучение процессов, происходящих при ударе молекул жидкости или газа о твердую поверхность, должно помочь выяснить природу К. 1) Удар о поверхность может произойти так, что молекула ориентируется, приближаясь к Ктр. одной своей частью (полярной) и отталкиваясь— другой. Ориентация может иметь место для молекул, обладающих электрич. моментом. Поскольку молекулы состоят из электронов и положительных ядер,возможна структура, при которой центры тяжести положительных и отрицательных зарядов не совпадают. Такие молекулы дипольны (смотрите Диполь мо-л екулярны й), то есть имеют электрический момент#0. При адсорбции молекула лежит на поверхности своей полярной группой. Такая ориентация может способствовать К. При дегидрогенизации голей последние прилегают к Ктр. своей группой СН2ОН, причем в этом месте происходит ослабление связей, активация и отщепление водорода. 2) Действие силового поля поверхности может вызвать деформацию адсорбированной молекулы, то есть смещение ее электрич. зарядов (поляризацию), в результате чего получается дополнительный электрический момент; такое нарушение нормального состояния способствует активации. Ориентационное и поляризационное взаимодействие может рассматриваться как проявление сил вторичной валентности. 3) При адсорбции незначительных количеств вещества может выделяться много энергии (экзотермичность адсорбции). Так, при поглощении углем первых порций кислорода выделяется 220 000 cal на г-молекулу 03. Это количество превышает теплоту горения твердого угля (96 000 cal) и близко к энергии горения газообразного углерода (около 150 000 cal), вычисляемой из теплоты его возгонки. Это указывает на то, что поверхностные атомы могут удерживаться слабо и быть в полусвободном состоянии. Выделяющееся тепло может служить источником активации. 4) Действие поверхности Ктр. может заключаться в ионизации. Легкость и быстрота взаимодействия ионов хорошо известны. При адсорбции кислорода на платине, повидимому, происходит переход электрона от металла на адсорбируемое вещество. Термоионный эффект (работа вырывания электронов) для платины равен 126 500 cal на з-атом; сродство молекул кислорода к электронам равно 80 500 cal. Если адсорбция заключается в ионизации кислорода за счет электрона платины, то требующаяся энергия составляет 46 000 cal, что близко к опытному инкременту энергии =й50 000 cal. Такое же соотношение наблюдается и при адсорбции на вольфраме. Ионизируются не все молекулы, ударяющиеся о поверхность Ктр., а только часть их. 5) Адсорбция на Ктр. может сопровождаться диссоциацией адсорбированного вещества на атомы; последние являются мощными активаторами. Тепловые эффекты реакций, если при расчетах исходить из атомов, значительно превышают энергию, выделяющуюся при взаимодействии молекул. Это объясняется значительной энергией, необходимой для диссоциации молекул (наир., Н2-»2Н— — 102 000 cal). 6) Силы, действующие между Ктр. и реагирующими молекулами, принципиально не отличимы от химич. сил. Вследствие неровностей и выступов поверхности Ктр., его поверхностные атомы не имеют окружения, соответствующего внутренним атомам, и могут рассматриваться как образования с ненасыщенными валентностями. При адсорбции могут образоваться временные «адсорбционные» соединения—комплексы, которые не всегда поддаются обнаружению химико-аналитич. путем, вследствие малой длительности их существования и быстрого перехода в устойчивые продукты реакции. Действие металлическ. Ктр. при реакциях с водородом и азотом может идти через образование гидридов и нитридов, существование которых доказано спектроскопии, методами. 7) Время пребывания молекулы в адсорбированном состоянии на поверхности Ктр. м. б. достаточно продолжительным. Это дает возможность другим молекулам, налетающим на поверхность, также вступать в реакцию. Такая фиксация молекул на Ктр. увеличивает число столкновений. 8) Специфичность действия Ктр. связана с его химич. структурой. Так, при бромировании этилена активными оказываются такие Ктр, у которых в состав молекулы входит кислород (стекло, стеариновая к-та, цетиловый голь и др.); на парафине скорость реакции в
17 раз меньше, чем на стекле. Введение про-пилового а на парафиновую поверхность при хлорировании приводит к возрастанию скорости. 9) В связи со сложным рельефом поверхности (трещины, впадины, «пики» атомов) и с активностью не всех, а только части общего числа адсорбированных молекул высказана теория, согласно которой на поверхности Ктр. активными являются только некоторые точки (активные центры), причем места, катализирующие одну реакцию, оказываются инертными для другой. Нагревание, связанное со скольжением атомов и с нивелированием поверхности, уменьшает число активных центров. На одних участках протекает химическая реакция, а на других—лишь обратимая адсорбция. При гидрогенизации этилена эффективна 0,01 поверхности никеля. Количество N молекул, налетающих в ск. на см2 поверхности Ктр., дается уравнением:
N=43,75 КГ6 -Λ., (10)
V мт где у—давление в барах (~10_6Him), Г— абсолютная темп-pa, М—молекулярный вес. Большое значение характера поверхности обусловливает тот факт, что скорость реакции в присутствии Ктр. не определяется однозначно величиной энергии активации, как видно из разложения Н-СООН на С02 и Н2, при различных Ктр. (табл. 2).
Таблица 2. — Энергия активации и к о н-станты скоростей разложения Н-СООН.
| Катализатор | Энергия активации в cal ыа г-молекулу Н-СООН | Константы скоростей при 200° |
| Стекло. | 24 500 | 2.5 · 10-7 |
| Золото. | 23 500 | 9,2 · 10-6 |
| Серебро. | 31 000 | 1,3 · 10-6 |
| Платина. | 22 000 | 4,4 · 10-4 |
| Родий. | 25 000 | 2.4 · 10-3 |
| Палладий. | 39 000 | 1.0 · 10-3 |
Действие платины и родия эффективнее, чем стекла и золота, хотя энергия активации остается того же порядка. 10) Ктр. оказывает специфич. влияние на ход реакции, обусловливая переход последней через промежуточные стадии. В связи с этим различные Етр. по-разному ускоряют одну и ту ate реакцию. Реакция образования С02 и Н2 из СО и Н20 протекает при 200—300° в присутствии меди; при 400—450° действует лучше окись железа. Причина—та. что в первом случае процесс идет через образование Н-СООН, а во втором—СО окисляется за счет окиси железа. Чередование окислительного и восстановительного процессов на поверхности Ктр. часто способствует сильному каталитйч. действию. Обусловливая неодинаковое течение промежуточных реакций, различные Ктр. могут приводить и к неодинаковым конечным продуктам. Разложение СН3-СО.ОС2Н5в зависимости от применяемого Ктр. протекает разными путями по ур-иям:
X. СНз-СО-ОСгН.-СНа-СО-ОН + С^Н, (Ктр.—ТЮ2);
II. СНз-СО-ОС2Н5=С,Н,+СО,(Ктр. — измепьч. Ni);
III. 2СН3-СО-ОСгН5=СНа-СО-СН, + Сг11а-ОН + СгН,-|-+С02 (Ктр.—ThOJ.
Точное экспериментальное определение энергии активации при гетерогенном К. затруднено вследствие изменения характера поверхности и степени адсорбции при изменениях ί°. В тех случаях, когда имеются основания полагать, что занята вся поверхность Ктр., энергия активации вычисляется из Г-ного коэфф-та. Как и в случае гомогенного К., активация может происходить за счет кинетич. или внутренней энергии молекул, ударяющихся о поверхность, за счет тепла адсорбции и т. д.
Изучение порядка реакции и влияния концентрации на ее скорость позволяет выяснить характер активации. Если для реакции необходимы молекулы А я В, то возможен случай, когда адсорбируется только молекула Л и процесс идет при ударе молекулы В о молекулу А, находящуюся уже в адсорбированном состоянии. Это—реакция первого порядка, определяемая количеством ударов молекулы В, поскольку адсорбционное равновесие между Ктр. и молекулой А устанавливается очень скоро (процесс не зависит от скорости адсорбции). Если обе молекулы, А и В, реагируют в адсорбированном состоянии, то скорость зависит от возрастающей концентрации, но когда последняя становится такой, что вся поверхность оказывается покрытой (то есть когда достигается предел адсорбции), дальнейшее повышение давления уже не меняет концентрации .молекул на поверхности Ктр. и не влияет больше на скорость, то есть реакция идет по ур-ию нулевого порядка. Этот случай имеет место при гидрогенизации на поверхности металлов, а также в нек-рых реакциях ферментативного характера.
К. в растворах зависит от растворителя, от прибавленных электролитов и от их ионов. Повидимому, в растворах происходит образование промежуточных комплексов. Скорость реакции зависит в этом случае не только от концентрации, но и от специально вводимых коэффициентов активности веществ, участвующих в реакции. Этими коэфф-тами выразится поправка на отклонение от законов идеальных газов в применении it растворам. Наряду с ускоряющим действием ионов установлено также и влияние недиссо-цшрованных молекул, причем ион и молекула характеризуются неодинаковыми константами скорости. Для случаев разбавленных растворов электролитов коэфф-ты активности вычисляются из законов электростатики и молекулярной статистики (закон Больцмана). Специфич. влияние оказывает на энергию активации и растворитель, как это видно на примере реакции между анилином и бромацетофеноном (энергия активации приведена в cal на г-моль).
Растворитель
Энергия активации
С„Н».. 8 000
CHCI».. 10 700
C.IlsNO,.. 13 4,70
О.Н»· CH.-OII. 14 290
С2Нб-ОН.. 13 900
Работа катализатора. Если V— объём газов, проходящих через Ктр. в ск., S—объём Ктр. (расчет ведется по объёму в виду невозможности количественно учесть величину поверхности), то выражение U —
V
= ^даст количество вещества, соприкасающегося с единицей объёма Ктр. в 1 ск. Если из проведенного через реакцию количества U прореагировала часть а, то выход будет равен αϋ. При небольших скоростях пропускания газов реакция успевает за время их соприкосновения с Ктр. дойти до состояния равновесия. С возрастанием U растет до некоторого преде- αίла и αϋ, поскольку длительность контакта оказывается достаточной и величина а—постоянна. На фигуре 1 (ось абсцисс—
U; ось ординат—αϋ) эта часть соответствует отрезку Оа. Точка а соответствует максимальному выходу при статических" условиях реакции, когда отсутствует циркуляция газов. При дальнейшем росте U начинается падение а, так как вследствие увеличивающейся скорости потока газов через катализатор успевает прореагировать меньшая часть вещества. Выход αϋ все же возрастает, так как уменьшение а компенсируется увеличением U (отрезок ab). Точка"?) указывает наибольший выход при динамическом проведении реакции (при непрерывном потоке газов). В случае еще больших значений U время контакта становится столь малым, что величина а быстро падает. Это ведет к уменьшению выхода (отрезок be). Слишком медленное пропускание газов через Ктр. практически неудобно; в первые моменты соприкосновения реакция протекает быстро, и дальнейшее задерживание газов над Ктр. мало эффективно. Напр., при продолжительности контакта МН3 и 02с платиной в течение 0,05 ск. выход достигает 96,1%; увеличение времени контакта в 12 раз (0,6 ск.) дает лишь незначительный прирост выхода в 0,2%. Поэтому технически оказывается выгодным, в известных пределах, увеличивать скорость протекания реакционной газовой смеси, не взирая на проистекающее от этого незначительное падение % выходов, но увеличивая зато нагрузку Ктр. Скорость газового потока нередко устанавливают в несколько м/ск. Необходимо также добиваться равномерного наполнения реакционного пространства катализатором во избежание образования свободных каналов, через которые газы могли бы проникнуть, не вступая в соприкосновение с ним. Состав пропускаемой газовой смеси определяется различными условиями, в зависимости от стоимости того или иного компонента и от влияния его избытка на конечный результат реакции. В тех случаях, когда реакция сильно экзотермична, но когда при этом повышение ί° выше определенного предела нежелательно (как, наприм., при контактном окислении аммиака), к смеси реагирующих газов примешивают такой избыток воздуха или другого газа, который поглощал бы излишнюю теплоту реакции. Зная тепловой эффект Q реакции и теплоемкости с всех
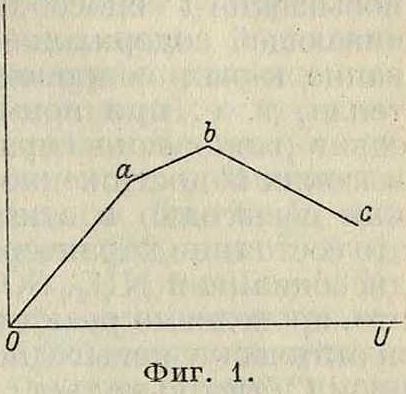
составных частей, подбирают такой состав газовой смеси, чтобы отношение ,Уе не пре восходило желательной (°.
К. и температура. Практически реакция ведется при t° наибольшего выхода. В экзотермич. контактных реакциях типа:
N,+ 3HS=2NH,
2S02 4- 02=2 S032NH, + ‘/.0,=2NO + 3H.0 4HCI + Оз — 2 Н20 -г 2 Cia Н,+-С1,=2НС1
+ 22000 cal + 45200 cal, + 107300 cal, + 29400 cal, + 44000 cal
повышение t° способствует реакции, увеличивающей содержание тех веществ, образо вание которых сопровождается поглощением тепла, то есть при повышении t° получается сдвиг равновесия справа налево. Т. о., при высоких Г достижение равновесного состояния невыгодно в отношении выхода, т. к. это состояние характеризуется значительной диссоциацией NH3, S03 и т. д. Несмотря на это, проведение реакций при низких t° тоже практически невыгодно, так как с понижением 1° быстро падает скорость процесса (смотрите Кинетика химическая); поэтому в последнем случае хотя и могут быть достигнуты большие выходы (вычисленные из константы равновесия), но самый ход реакции будет крайне медленным, аналитически неопределимым. Таким образом, ί° реакции не должен быть ни слишком высокой (что связано с непроизводительным распадом образующегося полезного продукта) ни слишком низкой. Практически при о г° проведении реакции в фигура 2. ограниченное время вы ход при низких ί° мал; далее, с ростом t° он увеличивается, достигая максимума, и при дальнейшем повышении t° снова падает вследствие неблагоприятных условий равновесия (увеличение диссоциации). На фигуре 2 по оси абсцисс отложена t°, по оси ординат—% выхода NH, при синтезе из N2 и Н2 в заданной начальной смеси. Кривая А дает концентрацию NH3 при равновесии как функцию ί°; кривая В—выход при наличии Ктр. Максимум с показывает оптимальную t° наибольшего выхода. Если Ктр. может с одним из реагирующих веществ давать соединение определен, состава, то i°-Hbie условия зависят от равновесия между Ктр. и данным веществом. К такого рода случаям относится контактное окисление S02 в присутствии окислов, характеризуемых более высокой оптимальной ί° действия, чем платина (смотрите Серная кислота). Напр., присутствие Fe203 в качестве Ктр. ведет к образованию сульфата Fe2(S04)3, к-рый находится в равновесии с S03 (продуктом своего распада), S02 и 02; каждой t° отвечает своя упругость пара S03 над твердым Fe2(S04)3. Проведение реакции с избытком SCX, в газовой смеси способствует образованию сульфата; при высоких Г повышается диссоциация S03, упругость паров последнего становится незначительной, и сульфат распадается. Оптимальной является та t° реакции, при которой концентрация S03 в равновесной смеси отвечает упругости паров
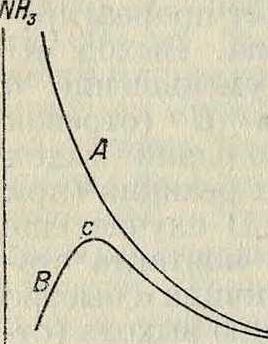
S03 над Fe2(S04)3. При этих условиях действие Ктр. может мыслиться как непрерывное чередование 2 процессов: образования связанного с Ктр. S03, затем отделения его и ухода из сферы реакции. Этими обстоятельствами объясняется то, что при начальном составе смеси из 7% SO2+10% 02+83% N2оптимальная <° каталитического действия Fe203 лежит около 625°, то есть выше оптимума для платины. В связи с этим изучение диссоциации гидридов, нитридов и других соединений должно дать возможность предвидения соответствующих условий К.
Активаторы (ускорители, промо-теры); смешанные катализаторы (впервые открытые В. Ипатьевым). При одновременном действии двух Ктр. общий эффект иногда бывает равен сумме эффектов каждого Ктр. в отдельности (влияние золей Pt и Au на распад Н202), но в большинстве случаев такой аддитивности не наблюдается: два или несколько Ктр. могут при совместном действии дать эффект, иногда превышающий сумму эффектов тех же Ктр., но взятых в отдельности. Вещества, присутствие которых усиливает уже проявляющееся каталитич. действие, называются активаторами, или промотерами (ргогпо-teurs); в качестве таковых применяются различные металлы, их окислы и другие вещества. Влияние промотеров нередко сказывается на скорости реакции даже тогда, когда они присутствуют в следах, едва обнаруживаемых химии. анализом. Промотеры, взятые в отдельности, могут и не проявлять того каталитич. действия, к-рое присуще основному Ктр. При введении в Ктр. промотеров в б. или м. значительн. количествах получаются т. н. смешанные катализаторы. Промотеры и смешанные Ктр. находят широкое применение в технике, где они дают возможность увеличивать выходы, комбинируя дешевые, легко доступные вещества. Примером промотеров являются примеси к Ктр.—железу при окислении NH3: при работе с чистым Fe выход равен 83,5%; Fe+Bi дает 94,6%; Fe+Cu 92%; Fe + Ce 90%; Fe+ +W 89,3%; Fe + Th 87,3%. С увеличением концентрациипромотера выходы растут,проходят через максимум и затем падают. Если Ктр. лежит на промотере, то малые количества последнего значительно влияют на скорость реакции. Если Ктр. и промотер равномерно распределены на подкладке,—влияние концентрации сказывается слабее. Возрастание константы скорости в процессе реакции может означать, что образующиеся вещества сами проявляют каталит. действие (автокатализ). При этом возможно возникновение ступенчатых реакций, протекающих с неодинаковой скоростью. Прибавление продукта, лишь медленно образующегося в начальной стадии реакции, равно как и введение дополнительного Ктр., ускоряющего одну из промежуточных реакций, протекающую с малой скоростью, дает увеличение выхода. Ускорение м. б. вызвано также посторонними газами, молекулы к-рых, сталкиваясь с активированными продуктами реакции, снимают с них избыточную энергию, превращая их в нормальные конечные продукты“ Такие случаи имеют место при тройных столкновениях, когда две реагирующие частицы легче взаимодействуют, если в момент их столкновения присутствует третья молекула, ноглощающая выделенную энергию и этим предотвращающая обратную реакцию.
Причины активирующего действия про-мотеров многообразны. 1) Если К. вызывается веществом А, то введение тела В, на поверхности которого А способно адсорбироваться, усиливает эффект. 2) Смешанный Ктр. может оказывать селективное действие, причем процесс, начинающийся на первом Ктр., завершается на втором. Напр., при реакции между СО и Н2, приводящей к образованию метана, Ктр.—никель дает промежуточное вещество, СН3ОН, к-рое дегидратируется на промотере (СН30Н->Н20-]-Н2СО> после чего из групп СН2 и Н2 образуется конечный продукт. 3) Промотер может ускорять образование промежуточного продукта, разлагаемого затем катализатором. 4) Соединение промотера с одним из реагирующих веществ или адсорбция последнего промо-тером увеличивают концентрацию данного вещества на поверхности. Зависимость между возрастанием коэффициента адсорбции и скоростью реакции сложна и мало изучена; известны случаи, когда в присутствии промотера адсорбция увеличивается на 20%, скорость же возрастает в десятки раз. 5) Действие промотера может заключаться в уменьшении чувствительности Ктр. к ядам (смотрите ниже), которые вызывают остановку реакции. 6) Ктр. и промотер могут активировать не одни и те же компоненты реакции; в известных случаях возможна и последовательная активация. 7) Подкладка может служить промотером, поглощая реагирующие вещества и являясь резервуаром, из которого Ктр. черпает материал для реакции.
Замедлители (ингибиторы); каталитические яды. Незначительные примеси нек-рых веществ либо замедляют реакцию (ингибиторы) либо практически прекращают ее (каталитические яды). Наличие 0,00002% РН3 лишает платину каталитич. свойств при окислении аммиака; 5-10~8 г-мол. ИСК уменьшают скорость распада Ы202вдвое. Различают яды временные и постоянные. Первые действуют лишь тогда, когда они присутствуют в реагирующих веществах; в этом случае продувание через Ктр. воздуха или другого инертного газа восстанавливает его действие. Таковы СО на платине при реакции между Н2 и 02 Или следы Н20 при синтезе Ш13 из элементов. Постоянные яды могут химически видоизменять поверхность, Ктр., причем для регенерации последнего требуется уже химич. обработка. H2S отравляет Ктр.—железо, образуя с ним FeS. Один и тот же яд м. б. постоянным или временным в зависимости от природы Ктр. На поверхности, отравленной для одной реакции, может протекать другая. CS2 отравляет коллоидальную платину, служащую Ктр. при гидрогенизации дипропилкетона, но не влияет на некоторые другие реакции, катализируемые той же платиной. Замедлителями м. б. и сами продукты реакции, адсорбируемые и задерживаемые на Ктр. Так, при окислении S02 на платине обра зующийся серный ангидрид S03 замедляет реакцию, так как последняя ставится в зависимость от скорости диффузии реагирующих веществ через слой S03, лежащий на платине и мешающий их проникновению к поверхности Ктр. Иногда примеси, уменьшающие выход, все же не являются ядами; так, ядами иногда называют вещества, которые сами действуют в качестве Ктр., но с той разницей, что они дают меньший выход или характеризуются более высокой оптимальной ί° сравнительно с обычно употребляемыми Ктр. Это относится, например, к у, к-рый считают ядом при контактном получении S03 с Ктр.—платиной; в действительности же As205 для данной реакции сам является катализатором, но с пониженным (по сравнению с Pt) выходом. Для Fe203 при 625° выход S03 составляет 75%; для FeAs04при 650°—63% и для As205 при 675°—50%. Ядом для контактного процесса является вода, которая с S03 дает H2S04, адсорбируемую на поверхности Ктр. Это имеет место и при высокой ί° в согласии с тем фактом, что небольшие количества адсорбированных веществ очень прочно удерживаются поверхностью. Создание условий, благоприятных для обратной реакции, вызывает замедление прямого процесса.
Действие ядов заключается гл. обр. в их адсорбции на поверхности и в уменьшении числа активных центров Ктр.Иногда присутствие яда может повлечь за собой даже изменение хода реакции. В связи с этим встречаются случаи полезного отравления, когда яд мешает идти побочной, нежелательной реакции. Наприм., при разложении а С2Н5-ОН на меди присутствие воды увеличивает выход альдегида СН3-СНО; безводный же дает больше побочных газообразных продуктов (СН4, С02 и СО) в качестве примесей к водороду. Объясняется это тем, что в безводном е идет побочная реакция: СН3 СНО^СН4+СО; вода, являясь ядом для этой реакции, защищает образующийся альдегид от распада.
Причины замедляющего влияния ингибиторов м. б. различными. 1) Роль замедлителя (А) при реакции в растворе может сводиться к уменьшению концентрации Ктр. (В), если происходит образование комплекса по ур-ию А+В^АВ и устанавливается равновесие между А, В и АВ. Тогда увеличение концентрации А вызывает уменьшение концентрации В (по закону действия масс). Таково, например, действие воды при этерификации к-т в гольных растворах или при разложении щавелевой кислоты водными растворами H2S04.2) Замедлитель может уменьшать концентрацию реагирующего вещества. При наличии иона в качестве реагента прибавление веществ, уменьшающих диссоциацию, действует замедляющим образом. Таково влияние НВг на гидролиз бромян-тарной кислоты. В ряде случаев, когда само образующееся вещество действует замедляющим образом (а в т о з а м е д л е-н и е),—скорость реакции обратно пропорциональна концентрации возникшего вещества и выражается ур-ием dx _ &г(а - х) di ~×5
Ύ. Э. т. IX.
30
где а—начальная концентрация, х—прореагировавшее количество ко времени t и — константа. Примером может служить действие НС1, HBr, HJ на гидролиз галоидозамещенных кислот. 3) В случае цепных реакций разрыв цепи может вызвать замедление; так же действует и дезактивация промежуточных продуктов. Образование НС1 из Н2 и С12 на свету идет так, что на один квант поглощенного света образуется 10° молекул НС1. Это—цепная реакция, вызванная первичным возбуждением, к-рое затем передается последовательной активацией;
С1 + 112 - НС1 + Η; Н + С12=НС1 + С1, и т. д. Введение 02, к-рый активируется хлором и дает с водородом Н20, разрывает цепь; аналогично действуют NH3 и нек-рые органич. вещества. Малые количества замедлителей могут оказывать очень большое влияние; наир., 1 мол. гидрохинона на 40 000 мол. акролеина делает аутооксидацию, автоокислс-иие (смотрите), последнего незначительной. Возможно, что этот процесс идет через образование перекисей соответствующих веществ
A. и последующее восстановление их при помощи вещества В, согласно ур-иям:
А + 0,=А(0,); А(02) + В=А(0) + В(0);
А(0) + В(0)=А + В + 0г.
Сопряженные реакции (смотрите), заключающиеся в том, что реакция между веществами 4иС, идущая медленно, ускоряется при одновременном протекании реакции между А и В,—представляют со стороны химичеек. кинетики аналогию с явлениями К. и автокатализа. Так как механизм сопряженных реакций обычно может быть прослежен, то изучение их дает косвенные указания относительно механизма реакций гомогенного катализа. я. сыркин.
Катализ в технике.
Каталитич. реакции играют в настоящее время весьма большую роль как в науке, так и в технике. Значительная часть процессов химич. промышленности, заводских и лабораторных синтезов базируется именно на каталитич. реакциях. Т. к. применение катализаторов (Ктр.) позволяет осуществлять химич. превращения с достаточной скоростью при ί° более низких,чем в отсутствии Ктр., то это дает экономию в топливе, а кроме того, нередко способствует повышению выхода и чистоты получаемого продукта.
Начало 20 в ознаменовалось важными открытиями в области каталитич. реакций, в скором времени обратившими на себя внимание промышленных кругов. Исследования в области катализа, предпринятые одновременно и независимо друг от друга во Франции Сабатье и Сандереном и в России
B. Ипатьевым, начиная с 1901 г., обогатили науку такими новыми методами, которые в скором времени произвели переворот в химич. технологии как органических, так и минеральных веществ. В связи с этим многие старые методы производства оказались невыгодными и подверглись коренным изменениям; многие каталитич. реакции, известные уже давно, нашли себе широкое применение в технике (синтез аммиака из элементов, поверхностное горение, окисление амми ака в азотную к-ту и т. д.), но главным стимулом развития каталитич. индустрии явилась война 1914—18 гг., когда Германия была отрезана от внешних ресурсов и сумела в крупном масштабе наладить каталитическое производство аммиака, серной и азотной кислот, отверждение жиров, и прочие Послевоенный период дал в области технического К. ряд новых блестящих достижений.
Катализаторы. Различные Ктр. обладают различной активностью, к-рую можно выразить некоторой величиной К, показывающей, во сколько раз один Ктр. трансформирует энергии больше, чем другой. Ипатьев выводит величину К на основании своих опытов со ами. Он считает, что если идет разложение в бомбе при нагревании, то простая пропорциональность р=р0(1 + «0 нарушается, и нарастание давления будет иттм пропорционально Т. Самая характерна и величина, определяющая ход разложения, есть, так как в это время посте-
ронние факторы влияют меиее всего. Если скорость разложения уменьшится, то время,
в течение которого достигается, увели-
/max
ЧИТСЯ, Т. е.
Величина Н изменяется для ов в зависимости от Ктр.; для Fe она почти вдвое больше, чем для А1203. Если R—константа одного Ктр., а —другого, то П6 — К. где
At 1
К—величина, указывающая на активность катализатора.
А к т и в и о с т ь Ктр. зависит в значительной степени от содержащихся в нем (или на его поверхности) примесей. Последние делятся на две группы. 1) Ли т и к а т а-лйзаторы (замедлители, каталитич. яды) ослабляют активность Ктр. или сводят ее к нулю («отравление» Ктр.). Типичными каталитическими ядами являются: галоиды, соединения серы, фосфора, а, HCN, СО и т. д. Так как ничтожной примеси этих веществ достаточно, чтобы парализовать действие Ктр., то материалы, подвергаемые контактным реакциям, должны быть совершенно чистыми, что значительно удорожает производство: наир., при синтезе аммиака по Габеру расходы по очистке водорода составляют 75% всей стоимости аммиака. В нек-рых случаях отравленный Ктр. можно «оживить» действием высокой t° или веществ, переводящих антикатализатор в летучее или индифферентное к Ктр. вещество. Отравленный Ктр. иногда пригоден для другой реакции. Так, Розенмунд, получая альдегиды из хлорангидридов, путем прибавления антикатализаторов к металлич. Ктр. выработал условия, при которых он мог по желанию останавливаться на любой стадии реакции:
C.HsCOCl -> Cell, сон ->С,Н,СН20Н -»с.н, · сн2.
2) Активаторы (промотеры, акцелера-торы) повышают активность Ктр., а следовательно, и скорость реакции. Например, равновесие
- т2 + з н2:5 2 NHs
в присутствии Fe достигается медленно, но прибавление к последнему ничтожных ко личеств Mo. W или U значительно ускоряет реакцию. Ипатьев, впервые обративший внимание на это явление, нашел. что амилен в медной трубке с СиО при 300° и 200 atm дает 30% пентана через 28 час., в то время как в железной трубке с СиО пентан образуется количественно через 12 час. Современная техника широко пользуется промоторами, так как они делают каталитич. процессы еще более экономичными.
Подбор пар Ктр. должен был бы производиться по определенным признакам, но это вопрос будущего; теперь они подбираются экспериментально. Особенно много проб со смешанными Ктр. было сделано при синтезе .метилового а из водяного газа (смотрите ниже), где применялись смеси из двух, трех и четырех компонентов.
Все огромное количество применяемых Ктр. можно разбить на следующие группы: 1) Ктр. для окисления—Pt, Rh, Ir, Pd, Os, Au, Ag, Fe203, Cr203, Mn02, CuO, Ni303, Co203, U02, V2Oj, W03, соединения Mo, редкие земли; 2) Ктр. для восстановления— металлы группы платины, Ш, Со, Си, Fe и их окислы, а также все Ктр., применяемые для дегидрирования; 3) дегидратирующие Ктр.—ΤΜ32, А1203, W03, алюмосиликаты, окиси металлов Cr, Si, Ti, Be, Zr, U, Mo, Fe, Ni, Zn, ZnCl2, к-ты, ангидриды и т. д.; 1) конденсирующие Ктр.—А1С13, FeCl3, CrCJ3, TiCl4, SnCl4, SbCl5, A1203, MgCl2, ZnCl2, CaCl, NH3, уксусный ангидрид, органич. основания и т. д.; 5) переносчики галоидов—
J, JC1, S, древесный и костяной уголь, хлориды Mo, Р, Sb, Fe, Sn, ΤΙ, V, U, AI, Zn. Переносчиками брома и иода являются бромиды и иодиды металлов, обладающих переменной валентностью.
Классификация каталитических реакций. Все огромное количество каталитич. реакций неорганической и органической химии удобнее всего классифицируется по след, схеме·: 1) окислительный К., 2) восстановительный
K. (гидрогенизация), 3) дегидрогенизация, 4) гидратация, 5) дегидратация и расщепление, 6) конденсация и полимеризация, 7) га-лоидирование, 8) отрицательный катализ.
1) Окнслительн ы и К. Процессы окисления, осуществляемые с помощью Ктр., подразделяются на две группы: а) окисление свободным кислородом или воздухом, б) окисление посредством окислителей, содержащих связанный кислород.
Процесс Дикон а—получение хлора из НС1 при помощи каталитич. действия СиС12. При этом процессе необходимо, чтобы НС1 не содержал паров серной к-ты, которая действует губительно на Ктр. Реакция ведется при ί° >400°; газообразный НС1 в смеси с воздухом проводится через башни, наполненные битым кирпичом, пропитанным раствором CuCl 2. Процесс Рейх л ера и Уайльда аналогичен дикоиовскому, но вместо СиС12 Ктр. служит сухая смесь MgCl2, MgS04 и МпС12.
Процессы получения камерной серной кислоты и олеума — окисление сернистого газа в серный ангидрид при помощи окислов азота или платинового и других Ктр.—также относятся к окислительным каталитич. реакциям (смотрите Серная кислота).
Процесс окисления аммиака в азотную кислоту, разработанный теоретически В. Оствальдом, имеет большое научное значение и широкое применение в технике. Имеется несколько технич. методов ведения этого процесса. А) М е то д Кайзера: Ктр. состоит из 4 отдельных слоев сетки из сплава Pt с небольшими количествами Pd и 1г. Кайзер первым стал применять Pt в виде сетки, т. к. это увеличивает поверхность контакта и тем самым повышает выходы. По методу Кайзера работал з-д в Шпан-дау (Германия). В России И. И. Андреевым, с нек-рыми изменениями в методах, был построен такой же з-д около Харькова. Б) М е-тод Франка и Каро: смесь воздуха с 12,5% NH3 подогревается в специальных подогревателях до 200°, после чего пропускается над Ктр. при темп-ре ~ 800°. Ктр. служит платиновая сетка (1 000 отверстий на 1 см2) общего веса 333 г. Вследствие чистоты поступающих газов активность Ктр. не уменьшается даже после полу года работы. В) Способ фирмы Байер (в Леверкузене) значительно отличается от предыдущих. Реакция ведется при 700—800°; Ктр. являются неблагородные металлы; точный состав Ктр. неизвестен; он состоит из Бе203с промотерами (вероятно, Сг203 и Мп203) и BiCl3 в виде гранул. Выходы по этому способу несколько ниже и достигают 80—85%. Камерные газы при описанных процессах заключают в себе около 10% N0, которую окисляют воздухом в Х02изатем действием воды переводят в к-ты:
2 N0 + 02=2 N0“; 2 N021- НаО=HNOg + IINO»;
3HNOg= HNOs + 2 NO + HaO.
T. к. полного поглощения полученных окислов азота водой достигнуть нельзя, то применяется последующее поглощение щелочью, которая превращает остаток окислов в смесь нитрита и нитрата:
2 N0, + 2 КОН -> KNOa + KNO* + Н.О.
Поверхностное горение сопряжено с полным окислением молекулы горящего тела. Разработка этого явления принад-лежит Bone, McCourt и др. (1902—14 гг.) в Англии, а также Шнабелю в Германии, которые применили этот принцип для технич. целей. Если какой-либо горючий газ в смеси с воздухом, взятым в количестве, достаточном для полного окисления, продувать че-ι рез пористую пластинку из огнеупорного материала, то после зажигания газа, прошедшего через поры, происходит ровное беспламенное горение в поверхностном слое пластинки, и последняя раскаляется добела. Ускорение сгорания зависит от физич. условий и от химической природы поверхности. Степень ускорения сгорания растет с повышением t° поверхности, так что поверхности, мало активные в холодном состоянии, при разогревании дают эффект, равный действию высокоактивной поверхности. Ускорение поверхностного горения зависит от адсорбции (окклюзии) горючего газа, причем горение происходит не во всей массе, но в слое глубиной не более 4—7 миллиметров. Во время горения пластинка электрически заряжается вследствие потери электронов раскаленной массой. На принципе поверхностного горения построено много приборов,обладающих большой экономичностью: кухонные плиты, печи для нагрева реторт и тиглей, трубы для нагрева паровых котлов. В последнее время появились приборы поверхностного горения, где пористая пластинка заменена пористыми шарами различного диаметра, что еще удобнее. Паровые котлы поверхностного горения сконструированы так, что трубы котла набиты кусками огнеупорного материала (кальцинированная магнезия или карборунд). Котлы эти были установлены впервые на заводах Skiningrove Iron Works в 1911 году и имели кпд ок. 90%. На принципе поверхностного горения и каталитич. действия поверхности основаны также друммон-дов свет, горелки Ауэра и т. д. Каталитич. реакции поверхностного окисления нашли применение и в военном деле, для перевода СО в СО г (смотрите Гопкалит, Противогазы).
Частичное окисление применяется гл. обр. в органических синтезах; при его помощи разрешается проблема превращения углеводородов в более ценные соединения— ы, кетоны, альдегиды и к-ты. Методика окисления в общих чертах заключается в проведении паров окисляемого тела, в смеси с воздухом, через слой Ктр. при нагревании. Непредельные углеводороды (олефины) при подобном окислении в присутствии W03 и Мо03 дают формальдегид:
СН2 : СН2 + Оа=2 Н СНО.
Ацетилен с АиС13 в водном растворе окисляется в глиоксаль и далее в щавелевую кислоту, с выходами до 80—85%:
•СН : СН + 2 0,=(СООЫ)а.
Довольно крупное технич. значение имеет окисление высших парафиновых углеводородов в к-ты. Для этого процесса используют твердый парафин, который окисляют сжатым воздухом при нагревании в присутствии раствора соды. Ароматич. углеводороды в присутствии Ктр. (лучшие: V205, Мо03, А1203) дают разнообразные продукты окисления. Η. Е. Орлов и другие получали над платиной из а бензальдегид, из ксилолов—смесь метилбензальдегида, фталаль-дегида и различных к-т; о-крезол при окислении над металлич. Ктр. образует салициловый альдегид и салициловую к-ту:
(Г°н -> Рг°> - Ггон
V—сн, ° н V"00011
Многочисленные патенты выданы на окисление нафталина или тетралина (тетрагидронафталина) во фталевый ангидрид:
ггсо
+ 90= О + 2 С02 + 2 Н20 + 449 100 cal
нафталин
Большое технич. значение имеет окисление антрацена в антрахинон при 450° в присутствии У205 или В(ОН)3 и Н3Р04 с 2% воздуха; выход доходит до 80%. При более энергичном окислении ароматич. углеводороды расщепляются, Бензол при постепенном окислении дает последовательно хинон и малеиновый ангидрид, к-рый далее распадается с образованием С02 и Н20. Каталитическим окислением ов можно получать альдегиды, кетоны и к-ты. Первичные ы жирного ряда с Ктр. Ag дают 70—90% альдегидов, вторичные ы окисляются труднее, но выходы кетонов достигают 90%. Третичные ы в этих же условиях разлагаются на формальдегид, кетон и воду:
у,0
(СНа),СОН + Оα= Н · СГ + СН3 · СО · СН, + НаО.
чн
Ароматич. ы при окислении над платиновой чернью дают альдегиды: бензойный, коричный и т. д. Окислением изоэвгенола в технике получают ванилин. Борнеол с Си при 200° дает 80% камфоры. Далее, альдегиды и кетоны легко переводятся каталитическим окислением в кислоты. При окислении карбоновых кислот чаще всего отщепляется С02 с образованием низших кислот, например:
СН, соон + зо=со, + н2о + н · соон.
2) Гидрогенизация (гидрирование). Эти реакции известны довольно давно, но всестороннее изучение их, выработка методики и приложение к технике относятся к успехам новейшей химии (смотрите Гидрирование). Способы гидрогенизации без давления разработали Сабатье, Сандерен, Майль, Мю-ра, Броше, Вильштеттер, Пааль, Скита и др. В России Ипатьев создал совершенно особый метод гидрогенизации под давлением. Гидрогенизация без давления заключается в пропускании паров гидрируемого вещества вместе”с водородом над Ктр. Согласно Сабатье, Ктр. для этой цели являются Ni, Со, Pt, Fe и Си (расположены в порядке уменьшения активности). Гидрогенизация ведется при различных темп-pax, в зависимости от вещества, но Г выше 300—350° мало применимы, т. к. при этом выходы уменьшаются; особенно важна чистота водорода, т. к. Ктр. очень чувствителен даже к следам ядов. Вильштеттер разработал метод гидрирования с Pt- и Pd-черныо, внося их во взвешенном состоянии в раствор реагентов и взбалтывая, при одновременном пропускании водорода., Этот метод гидрогенизации очень точен и позволяет останавливаться на любой стадии восстановления. Впервые он был применен Фокиным при гидрировании олеиновой кислоты в стеариновую к-ту. Пааль и Скита применяют метод гидрирования, стоящий на грани между К. гомогенным и гетерогенным. Ктр. здесь служат коллоидные растворы Pt и Pd, полученные по Бре-дигу и стабилизованные защитными орга-нич. коллоидами; гидрогенизация ведется обычно в водных, водноовых или уксуснокислых растворах. Метод Келле позволяет применять коллоидные Os и 1г. Способ Броше занимает среднее место между гидрированием под давлением и без давления. Броше пользуется автоклавом с мешй; реагирующая смесь наливается в него на 2/3, вводится 5% Ктр. и водород под давлением 15—20 aim; водород добавляется по мере его израсходования.
Гидрогенизация под давлением (метод Ипатьева) производится в бомбе Ипатьева — специально сконструированном им аппарате для высоких давлений (рисунок см. в ст. Гидрирование). Последний состоит из прочной стальной цельнотянутой трубы (емкостью от 250 ом3 до 2 л), к которой болтами присоединяется головная часть с манометром и впускным вентилем. При достаточном количестве водорода гидрирование считается законченным, когда при постоянной t° давление перестает уменьшаться. Реакция протекает тем скорее, чем теснее соприкосновение реагентов со всей массой Ктр.; размешивание значительно ускоряет процесс. Ипатьев применяет для гидрирования в качестве Ктр. чаще всего NiO (металлические Nib этих условиях действует слабее), которая хорошо гидрирует ароматические кольца; для гидрирования ненасыщенных боковых цепей без гидрирования ядра применяется СиО.
При помощи гидрирования можно ввести водород в самые разнообразные оргаяич. соединения. Этиленовая связь при гидрировании легко превращается в простую. Этилен при полном гидрировании дает этан, но при недостатке водорода получаются масла, по свойствам близкие к нефти. Гидрогенизация но месту двойной связи имеет огромное промышленное значение в процессе отверждения жиров (смотрите Гидрогенизация оюиров). Первое технич. осуществление этого процесса (с Ктр. Ni) было сделано Лепренсом и Се-веком в Германии (1900 г.) и Нордманом в Англии (1900 г.). В России Фокин успешно гидрировал жиры, применяя Pd- и Pt-чернь. Ипатьев (1907 год) произвел серию исследований с самыми разнообразными жирами и маслами; он нашел, что при 120° с Ni и при 250° с Ni203, под давлением всего лишь в 25—30 atm, гидрирование идет хорошо, причем жиры можно брать и неочищенные. Бедфорд и Эрдман (1909 год) предложили для технических целей NiO на кусках пемзы при 170—200°, с прибавлением разнообразных промотеров (Zn, Ti, Се, La, Mg и т. д.). Этим способом пользуется Баденская анилиновая ф-ка (BASF). Кроме этих способов, существует громадное количество патентов с предложением самых разнообразных Ктр., как то: разнообразные металлы, окислы и их смеси, никельсодержащие мыла, никелевые соли жирных к-т, ацетаты Fe, Ni, Со и других металлов, оксалаты, никель-карбонил, соли пикриновой к-ты. Лучшими Ктр. являются благородные металлы—Pt и Pd; например, 1,7 г Pt в течение 3 — 4 часов превращает 10 килограмм жира в твердое состояние. Очень интересным Ктр. для гидрирования на холоду (по Леману) является четырех-окись осмия, Os04.
При крекинге нефти (смотрите Крекинг-процесс) и других минеральных масел непредельные соединения, получающиеся при разрыве более сложных молекул, путем гидрирования можно превратить в предельные бензины. Из ароматич. углеводородов превращается в метилциклогексан, ксилол—в диметилциклогексан, дифенил—в ди-циклогексилметан и т. д. Все сполна гидрированные ароматич. углеводороды представляют собою в большинстве случаев жидкие масла, довольно инертные по отношению к кислотам и щелочам, почему они и находят применение в технике как хорошие растворители. Углеводороды с конденсированными циклами при гидрировании внедряют водород сначала в одно кольцо, затем в другое. Гидрированием нафталина без давления или, лучше, под давлением получают тетрагадро-нафталин и декагидронафталин, известные в технике под названиями тетралин и декалин. Тетралин в настоящее время получается тысячами т и находит разностороннее применение. Для получения его нафталин очищают от примеси сернистых соединений, перегоняют и гидрируют под давлением с никелевым Ктр. В 1928 г. был применен совершенно новый метод гидрирования углеводородов действием сплава натрия с калием (Na, К) и гидрюра натрия (NaH2). По этому способу 1 000 ч. нафталина в смеси с 70 ч. кизельгура, 27 ч. натрия и 93 ч. калия при 15 atm и 230° через 2 часа дают 95%-ный выход тетралина. Альдегиды и кетоны при гидрировании превращаются в первичные и вторичные ы (над никелем при 180°), причем кетоны редуцируются несколько труднее альдегидов:
Л>
R-C< 4- H2->R СН2ОН ;
ХН
R · СО · R + Н2-*В. · СН(ОН) · R. Формальдегид при этой реакции дает плохой выход метилового а, т. к. восстановление идет дальше до образования метана:
у,о
Н · Cf +2Н,^ сн“ + н2о. хн
Ипатьев при гидрировании альдегидов и кетонов под давлением 100 atm и 250° получал хорошие выходы соответствующих ов. Лучшими Ктр. являются NiO и СиО. Фенолы при гидрировании легко присоединяют водород, переходя в алициклические соединения; например, гидрирование обыкновенного фенола дает смесь циклогексанода и циклогексанона. Образование этих двух продуктов Ипатьев объясняет существованием кето-энолыюй таутомерии (смотрите) фенола. сн сн^ сн
СН^/СОН
сн
t I ! 1
, сн.
I
со сн2
н2с сиг
н2сЬ|снон сн2
циклогексанол сн2
сн2 сн.
СНа /С°
сн2
циклогексанон
Гораздо удобнее получать циклогексанол гидрированием салициловой к-ты, так как в этом случае ему не сопутствует циклогексанон, от которого очень трудно освободиться. При пропускании высших фенолов в токе водорода над Ni, Си, Ag, Pt, а особенно над ШСг04, происходит отщепление боковых цепей:
-он
-сн„
+ н2
-он
+ сн4
Т. о. из крезолов м. б. получен технически ценный фенол. Высшие фенолы, например ксиленолы, переводятся этой реакцией, в конечном итоге, тоже в фенол. Фенолы можно восстановить далее в углеводороды при пропускании их над углем с железом при 430°: у он
С,Н4( +C-+C.Hs-CHs + CO.
хси3
Фенолы можно превратить в углеводороды и обычным гидрированием. Первичные смолы (Uhrteer), которые получаются при перегонке углей при низких t°, дают превосходное тяжелое топливо для дизелей, но присутствие в нем фенолов крайне вредно для моторов. Вместо выделения фенолов гораздо рациональнее превращать их гидрированием в углеводороды, пригодные в качестве моторного топлива. Ф. Фишер редуцирует фенолы в смоле, пропуская последнюю через нагретую железную внутри луженую трубку в токе водорода или водяного газа. Нитросоединения, ароматические и жирные, при гидрировании дают хорошие выходы соответствующих аминов:
Е · N0* + 3 Н„ -> R · N11. + 2 НаО.
В случае нитробензола анилин получается лишь при малоактивном Ктр., т. к. он легко гидрируется далее в циклогексиламин. На способы получения анилина из нитробензола взято много патентов. Лучшими- являются способ Броуна и Генке (при 230° с Sn, SnO и Sn02) и способ Пома и Пеллегри-ни, которые ведут реакцию в смеси нитросоединения с водой при 4—5 atm и 50—60° с 0,5% Ктр. (Ni, Со, Си, Fe) на индифферентном субстрате из угля, пемзы или кварца.
Синтезы из во д я и о г о газа. Среди разнообразных синтезов, основанных на реакциях гидрирования, к-рыми располагает и пользуется современная химич. индустрия, важное место занимают синтезы из водяного газа и других газов, являющихся отбросами заводских процессов. При помощи Ктр. из малоценных газовых смесей удается получать такие ценные вещества, как жидкое моторное топливо, высшие ы и кетоны, углеводороды и т. д. Первые попытки соединения СО и Н2 в жидкие продукты производили Лозанич и Иовичич (1897 г.), подвергавшие смесь этих газов действию электрич. разрядов. При этом ими был получен формальдегид, к-рый конденсировался в маслянистые продукты. Ацетилен с СО в тех же условиях дает светлокоричневую твердую массу, этилен—желто-красное масло, а бензол—густую темную жидкость. Сабатье действием водорода на СО при 250° получил нацело метан; выше этой Г идет уже процесс разложения СО:
С0 + ЗНа=Н,0 + СН, (Г $ 250°);
2СО=СО, + С (Г > 250°).
Метай пробовали также получать прямым гидрированием угля; найдено, что без Ктр. эта реакция идет только при 1 200°, но в присутствии Ni и Со — при более низких ί°. Ипатьев из СО-(-Н2 при 510—525° под давлением получил лишь 6,5% СН4. Восстановление СО2 идет в первой фазе до СО, а затем приводит к образованию СН4. В настоящее время разработаны технич. методы получения СН4 из СО и С02 при помощи К.
Си и т е з ы из СО+ Н2. В 1908 г. Орлов получил из смеси СО+Н2 при 100° ненасыщенные углеводороды, пользуясь Pt и Pd на асбесте. Ф. Фишер и Тропш в 1913—14 гг.,
работая с водяным газом, содержащим избыток водорода, получили смесь углеродистых соединений, названную синто лом; эта смесь в качестве горючего с успехом может заменять керосин. Синто л получают при циркуляции газов под давлением 150 atm при410—450° над содержащим щелочь Ктр. Согласпо данным анализа, сиитол представляет собою сложную смесь, которая содержит: 29% голей, альдегидов и кетонов и 48% масла, летучего с паром, содержащего высшие ы, альдегиды и кетоны. Кроме того, в условиях опыта получения синтола образуется: 10% различных органич. к-т и 2% конденсатов, не летучих с паром. Син-тол—светложелтая жидкость, не темнеющая при стоянии, уд. в 0,8289, начинает застывать при —30°, окончательно затвердевает при —90°. Синтол, освобожденный отмыванием от органич. кислот, представляет собою топливо с теплотворн. способностью 7 500 8 200 Cal (керосин дает 11 000 Cal). Выход— 3 килограмма из 10 м3 газа. Авторы метода полагают, что во время реакции сначала получается растворимая в воде смесь ов, альдегидов, кетонов и кислот (иросинто л), которая при дальнейшей конденсации образует нерастворимый в воде синтол. Процесс образования синтола протекает, вероятно, по следующей схеме:
СО + 2Н2=СН,ОН;
CHjOH + СО=СНзСООИ ;
2 СН,СООН=СНзСОСНа + СОз + Н„0 ;
СНзСООН + Н.=СНзСОЙ + Н.0 ;
СН.СОН + Н,=СНзСН-ОН;
СНзСН.ОН + СО=СН,СНзСООН, ит. д.
При нагревании в автоклаве под давлением (в отсутствии СО) синтол отщепляет воду и переходит гл. обр. в нафтеновые углеводороды с ясно выраженным нефтяным запахом (вероятно, высшие толи переходят с отщеплением воды в олефины, а последние, с замыканием кольца,—в нафтены). Этот нефтеподобный продукт превращения синтола называется си н тин ом. Несомненно было бы значительным успехом, после экстракции из угля первичной смолы, газифицировать полукокс, от него переходить к водяному газу и из последнего получать синтол. Этот метод таит в себе большие перспективы для стран, богатых углем и бедных нефтью.
Синтез нефти. Фишер и Тропш действием различных Ктр. на водяной газ получили в небольших размерах синтетич. нефть. Авторы заметили, что применение в качестве Ктр. Fe и ZnO увеличивает количество нефтяных углеводородов, применениеже Fe203 и Сг203 дает смесь, богатую твердыми парафинами. Для реакции необходим Ктр., содержащий щелочь, так как щелочи значительно активируют процесс. Из большого числа испытанных Ктр. лучшими оказались Со и Pd; активаторами, повышающими выходы, являются ZnO, BeO, Mg(OH)2, Mn(OH)2, Cr203+Pd, Си, Fe203 и следы щелочей. Синтез ведется при 250° под нормальным давлением. В числе продуктов получаются: 1) газо ль, горючий газ, состоящий гл. обр. из смеси С3Н„, С3Н8, С4Н10; 2) синтетич. бензин (20%) с t°mm_ 30—170°, гораздо более чистый, чем пенсильванский; 3) керосинообразный продукт (22%) с t°KU,u 170—
330°; 4) чистый твердый парафин с t°n.,_ около 61°. Образование углеводородов авторы объясняют промежуточным возникновением карбидов; например, для Ктр. Со реакция м. б. представлена так:
СО + Н. + Со=СоС + Н.О ;
СоС + II.=Со + СН»; пСН.=(СНа)п;
(СН2)п + На=СН3(СН2)п_2СН,.
Способ этот, наравне с синтольным методом, вероятно, вскоре разовьется, т. к. при помощи его можно будет успешно превращать бесполезные топочные газы в жидкое топливо, являющееся пульсом современной промышленности.
Синтезы метилового а. Попытки получения метанола из водяного газа делались давно; первых положительных результатов достиг в 1921 году Кальверт (с выходами до 80%). Согласно Кальверту (подробного описания метода не имеется), водяной газ можно получать из всяких отбросов—коксовой и угольной пыли, древесных опилок ит.д., благодаря чему синтетич. метанол должен быть гораздо дешевле древесного. По патенту Леша (Lush), водяной газ быстро пропускается под давлением над Ктр., состоящим из 4 ч. Ni, 1 ч. Си и 5 ч. А1203; этот метод дает, однако, наряду с метанолом много формальдегида и продуктов его полимеризации. Наилучших результатов достиг Г. Патар: путем циркуляции водяного газа или смеси СО с избытком Н2 над Ктр. достигается полное превращение их в метанол: СО + 2Н3->СН3ОН + 27 000 cal.
.Восстановление идет лучше всего при 400— 420° под давлением 150—250 atm, в присутствии ZnO, осажденной на асбесте. Из 300м3 газа получается 100л 80%-ного метанола. Митташ, Пиер и Винклер на заводе BAMAG производили те же опыты, носдр. Ктр., состоящими из окислов металлов с разнообразными активаторами (Zn0 + Cr203, Zn0-bV205, Cd0 + Cr203, и т. д.). Им удалось добиться прекрасных результатов при давлении 50—100 atm и 400°. Для получения синтетич. метанола пет необходимости иметь чистый водяной газ. Варианты в составе исходной газовой смеси м. б. очень разнообразны, при условии отсутствия ката-литич. ядов; присутствие азота даже в избытке лишь несколько замедляет реакцию. В случае избытка СО или Н2 против стехиометрической пропорции синтез идет со скоростью, пропорциональной парциальным давлениям. Необходимыми условиями при синтезе метанола являются: 1) отсутствие в материалах аппаратуры Ni и Fe, каталитически превращающих в метан, и 2) полное отсутствие щелочности в Ктр., так как иначе наступает синтолообразОвание. Аппаратура изготовляется поэтому из меди, серебра, алюминия или их сплавов; для мелких частей применяют сплавы цинка, сурь-дш и свинца. Лучшим материалом являются специальные сорта стали: Y2A Круппа, ви-крометалл и т. д. Синтез метанола, являющийся новым производством, быстро развивается: в 1927 г. в Германии получали уже свыше 20 тонн метанола в сутки. В последнее время появилось много патентов на вариан ты синтеза Патара. Напр.,Вудфуд (Wood-food) и Блофилд (Blowfield) проводят газы при давлении 140 atm (2 000 фн/дм.2) при 385—420° над Ктр. из окисей Zn, Сг, Fe; особенно рекомендуется ими ZnCl2. Compagnie de B6thune применяется хромоникелевый Ктр. при давлении 800 atm и 300°; выход около 75%. Societe Frangaise de Catalyse Generalis6e применяется Ктр. из смеси Sr и Zn с РЬО или Bi203 при давлении 200 atm н 300°; выход метанола количественный.
Синтезы аммиака. Мировая азотная промышленность растет с каждым годом, благодаря значению ее главным образом для производства удобрений и чатых веществ. Все государства покровительствуют этой отрасли промышленности, т. к. природные запасы селитры истощаются и приходится переходить на синтетич. методы. Синтез азотной кислоты по Эйде и Биркеланду и синтез цианамида кальция по Франку и Каро завоевали себе прочное положение в промышленности, но сильным конкурентом их является синтетич. аммиак, получаемый непосредственно из элементов при помощи различных Ктр. (смотрите Аммиак). Остин (1788 г.), Деви (1800 г.), Генри (1809 г.) установили, что при пропускании электрич. искры через смесь трех объёмов водорода с одним объёмом азота образуется два объёма аммиака. Девиль (1865 г.) показал, что реакция n2 + з н. ^ 2 NH3
обратима. Лавати предложил синтезировать аммиак пропусканием смеси азота с водородом через сосуд, наполненный смесью титановой земли с платиной на пористом субстрате. Шарль Телье (псевдоним Dufregue) взял несколько патентов на синтетич. аммиак; он получал его в значительных количествах, пропуская смесь N2 и ЗН2 над губчатым железом при ярко красном калении, причем им были разработаны технич. детали с применением давления до 10 atm. Дю-Мотай (Tessie du Mottay) взял патент на синтез аммиака из элементов при помощи нитрида титана при ί° красного каления. Ле-Шателье вывел теоретически, что синтез ΝΗ3возможен лишь при высоком давлении в присутствии Ктр.; экспериментальные исследования его были прерваны случившимся ом. Честь детальной разработки технич.синтеза ΝΗ3 принадлежит Габеру, Неристу и Оордту, Ле-Россиньолю и Гринвуду. После долгих исследований и усовершенствований аппаратуры, с 1910 г., при поддержке BASF была осуществлена техн. эксплуатя способа Габера. Несмотря на различные затруднения, Габер при помощи д-ра Боша с блестящим успехом сумел поставить свой синтез в грандиозном масштабе. Первый з-д их появился в 1913 г.,в Оппау; в 1927г. заводы Леуна у Мерзебурга уже дала 340000 тонн NH3.
а) В процессе Габера и Боша смесь газов готовится по способу Боша, применяемому BASF: в газогенераторных печах, питаемых коксом, получается смесь СО и N2, которая смешивается с водяным газом, полученным по методу BAMAG; эту смесь пропускают с избытком водяного пара над Ктр. из активированной окиси железа. После поглощения полученного С02 адсорберами с водой под давлением смесь содержит N и Н
почти в стехиометрии, пропорции (N2:H2== 1:3 по объёму). После последующей очистки и сушки смесь поступает в контактные аппараты. Удаление СО (являющейся ядом для Ктр.) путем промывания газов аммиачным раствором закиси меди—неудовлетворительно, так как при этом отлагается медь и разъедается железная аппаратура. Несколько лучше действует записная аммиачно-медная соль муравьиной к-ты, но поглощение СО получается неполное. В Оппау газы пропускают для очистки через стальные бутыли, под давлением 200 atm; в первых восьми бутылях циркулирует указанный выше формиат меди, а в последующих—едкий натр при 260°. Для осушения газы пропускают над амидом натрия, NaNH2. Для предохранения Ктр. от возможности отравления СО очень рекомендуется ставить «протектор» с Ктр., чаще всего с Fe или Ni, при условиях, обычно применяемых для гидрирования. Синтез NH3 выполняется при давлении 200 atm и t° ок. 500°. В аммиак практически превращается 7—9% смеси на каждую контактную единицу; общий выход достигает 80% теории. Первоначально реакционные бомбы нагревались снаружи, но этот способ в настоящее время оставлен, т. к. водород при высокой ί° декарбонизует сталь. Новые типы аппаратов строятся из специальных сталей с малым содержанием углерода. Для начала реакция требует предварительного подогревания смеси или Ктр., после чего она поддерживается собственным теплом. Полученный аммиак сжижается в конденсаторах, а непрореагировавшая смесь возвращается в цикл. б) Способ Клода характерен «сверхдавлением» в 1000 atm, почему получается лучший выход, т. к. для 1 000 atm и 550° равновесие отвечает содержанию 41% NH3 (вместо 13% у Габера). Аппаратура построена из никельхрома или специальных сплавов «ATG», Yichronic. Оболочка катализаторной бомбы имеет толщину стенок 64 миллиметров и длину ок. 2 метров Трубка с Ктр. привинчена снаружи, так что ее можно переменить в 10 мин. Контактных аппаратов четыре: через первые два газ идет параллельно, через два последних— последовательно. Смесь нагнетается сверхкомпрессором при 1 000 atm и через подогреватели подходит к «протектору» с Ктр., который улавливает яды (гидрирует СО в СН4 и О в Н20). За протектором стоит холодильник, где конденсируются следы воды; чистые газы затем проходят через подогреватель, две первые бомбы с Ктр. и затем, после охлаждения и подогревания, через две последние бомбы. Непрореагировавшая смесь (ок. 20%) возвращается обратно, а полученный аммиак сжижается. Выход равняется 5 тонн сухого аммиака в день на 1 производственную единицу описанного типа. в) Способ Казале отличается от способа Габера применением больших давлений; при этом выход ΝΗ3 достигает 20%. В большинстве установок системы Казале пользуются электролитическим водородом, к-рый освобождают от примеси кислорода каталитическим сжиганием соответствующего количества 2Н2+02в воду. Процесс синтеза NH3ведется при 750—800 atm и 400°. Темп-ра
Ρΰτρ. поддерживается теплотой реакции после предварительного подогрева. Ктр. служит железо или окись его с различными активаторами (Со, Ni, Μη, Mo, W, U), а также c углем или окисями легких металлов (Mg, А1, Са), которые являются одновременно и субстратом и активатором. г) В способе Фаузера азот очищается от остатков кислорода и от примесей N0 пропусканием над медью при 400°. Смесь проходит при 250 aim и 500° через восемь контактных башен. Ктр.—окись железа с добавкой 4—5% активаторов. Продукция—до 10 тонн в день на производственную единицу. д) В способе Мон-Сени (Mont-Cenis) смесь нагнетается в Ктр. под давлением ок. 80 atm, причем 10—20% азота переводится в аммиак. Ктр.—железо с комплексными алюмо-железосинеродистыми солями (точный состав его держится в секрете), отличающийся особой активностью. Темп-ра реакции— около 400°. Так как и давление и температура реакции низки, то аппаратура может быть изготовлена из обычной сименс-мар-теновской стали. е) Способ Цедербурга (Norsk Hydro-Elec-trisk) применяет в качестве Ктр. комплексные ферроцианиды со щелочными металлами.
Сейчас известно, кроме перечисленных, еще много способов синтеза NH3, но все они являются только вариантами указанных. Углекислота, которая в процессе Габера и Боша при очистке газов поглощается в скрубберах (на 1 тонна NH3 получается 45 000 фт.8 С02), находит применение для получения аммонийных солей.
В настоящее время синтезы аммиака в мировой азотной промышленности занимают доминирующее место. Мировая добыча связанного азота по отдельным отраслям промышленности распределяется так (в %):
| 1913 г. | 1926 Г. | 1927/28 г. | |
| Связанный азот из | |||
| воздуха. | 7.1 | 55,8 | 55,9 |
| Аммиак из камснно- | |||
| го угля. | 36,7 | 23,6 | 16.5 |
| Чилийская селитра | 56,2 | 20,6 | 27,6 |
Принимая во внимание все строящиеся заводы, можно считать, что на 1930/31 г. будет получено синтетич. методами 2,2 млн. т связанного азота. Германия по синтезу аммиака занимает первое место, т. к. способ Габера и Боша в ней используется еще и для других целей, требующих высоких давлений: в Леуна и Оппау на габеровских установках работают з-ды искусственного бензина. В виду своей относительной простоты синтез аммиака в настоящее время обходится дешевле всех остальных методов получения связанного азота. Каро указывает, что цианамидный способ может идти наравне со способом Габера лишь при наличии дешевой электроэнергии. В настоящее время во всех странах промышленность синтетич. аммиака широко развертывается. Крупнейшими азотными синдикатами являются: I. G. в Германии, Азотный синдикат Каро и концерн Кульмана, во Франции, Ni-tram Ltd. в Англии, Dupon de Nemour в Соединен. Штатах «Америки, концерн Монте-катини в Италии. Продукция связанного азота из воздуха на 1927 г. составляла (в тоннах):
Германия. 450.000 Англия. 14 400
Франция. 43 000 Бельгия. 12 000
Италия. 34 500 Испания. 6 000
С. Ш. А. 32 450 Швейцария. 2 200
Япония. 27 000
3) Дегидрогенизация (дегидрирование). Для процессов дегидрогенизации Ктр. являются те же вещества, что и для гидрогенизации. Обычно дегидрогенизация наступает при более высокой έ°, чем гидрирование. Ипатьев, Сабатье и др. показали, что в случае альдегидов, кетонов и ов су-. ществует равновесие, когда скорости гидрогенизации и дегидрогенизации взаимно уравновешиваются. Дегидрогенизация органич. соединений с открытой цепью обычно протекает сложно, с образованием многочисленных продуктов, расщеплением цепей и выделением угля. При этом одновременно имеют место два основных процесса: а) распадение сложных молекул на более простые и б) полимеризация обрывков цепей в высокомолекулярные продукты. Разложение ацетилена над Ni, изученное Сабатье, приводит при 150° к образованию сложных керосино-подобных смесей жирных, жирно-ароматич. и ароматич. углеводородов.
Крекинг нефти. Дегидрогенизация имеет в технике большое значение для получения из высококипящих масел более низ-кокипящих продуктов разложения, которые заменяют бензины. Это было впервые отмечено в 1792 г. Мердоком, к-рый при нагревании тяжелого масла получил смесь горючих газов. Расщепление масел при крекинге доводится до образования лишь жидких продуктов; при дальнейшем повышении t° происходит дальнейшая дезаггрегация цепей, с образованием газов и угля. Последние процессы называются пирогене-з о м. Обычно крекингу (смотрите Крекинг-процесс) подвергают не сырую нефть, а нефтяные погоны—пиронафт, соляровые масла—с целью’получения из них бензинов. При 600— 900° наступает пирогенез с образованием газов; поэтому крекинг ведут при 300—600°, причем получается смесь предельных и непредельных углеводородов. В имеющихся условиях непредельные соединения обычно по-.тимеризуются и дают смолистые продукты, которые в свою очередь распадаются, давая в числе продуктов уголь и водород. Крекинг нередко ведут в присутствии Ктр.; Ипатьев нашел, что хорошим Ктр. для этой цели является глинозем. Ипатьев в 1907 году и Сабатье в 1909 году соединили крекинг с гидрогенизацией—для перевода непредельных углеводородов крекинга в предельные. Сабатье с Ni при 150° или с Fe при 400° получил до 75% предельного бензина. Шпиндлер в 1928 году описал интересный способ получения насыщенных и ненасыщенных цепных и кольчатых углеводородов путем пропускания этилена через различные Ктр. (Fe, Ni, Со, Си, Cr, V, Се, Pt, Pd) при 500° и давлении 1 000 atm. Действием пламенной дуги при 1 300° и 650 atm на СН4, С2Н6, С3Н8 и т. д. в присутствии Se, Те, Тй, силикагеля, активированного угля—эти углеводороды м. б. целиком разложены на ацетилен и водород.
Крекинг под давлением дает меньший % газов и смол и большие выходы бензина.
Крекинг бакинской нефти с t°mn. 360—420° под давлением 420—450 atm дает (из 500 л): 50—100 л газов (80% СД,-|-20% непредельных углеводородов), 320 л масла (90% с t0кин. Д° 280°) и 50 л пека. При новейших способах крекинга применяются невысокие давления порядка 10 — 15 atm при темп-ре 500—600°. В год крекируется ок. 10 млн. м3 нефтяных погонов. Введение вместе с нефтью паров воды уменьшает нагар и образование кокса, т. к. вода окисляет уголь в СО, давая водяной газ. Часто крекинг ведут с водородом и Ктр. в условиях, напоминающих бергинизацию (смотрите Жидкий уголь).
При крекинге углеводородов с открытыми цепями получаются всегда и кольчатые углеводороды. очень стойкие к нагреву. Ароматич. углеводороды в этих условиях уплотняются в многоядерные или конденсированные циклы. На этом принципе основаны заводские способы получения дифенила из бензола, дитолила из а, а также различные конденсации, связанные с дегидрогенизацией, например, переход дибензила в фенан-трен, дифенилметана во флуорен, и т. д. При помощи этого метода получаются трудно доступные: флавантрен, бензодиантроны, перилен, бензантрон, нафтодиантрон, виол-антрен, различные дериваты пирантронов и т. д., представляющие собою сложные системы из 5—10 конденсированных бензольных колец. Дегидрирование простых циклич. соединений с 1 — 2 шестичленными кольцами, насыщенными водородом, идет легко при пропускании их паров через трубку с Ктр. В качестве последнего особенно пригодны Ir, Rh, Pt, Pd. Обычно процесс идет несколько глубже, и, вследствие частичного разрушения колец, образуются также газы.
Исследование процессов дегидрирования с благородными металлами в качестве Ктр. принадлежит Сабатье и Сандерену, которые вели реакцию при 400° и не наблюдали при этом распада бензольных колец. Зелинский на основании опытов Ипатьева применил более доступный Ктр.—Ni(N03)3 с алюминатом натрия [или А1202(0Н)2 с 50% Ni]; дегидрогенизация идет количественно без разложения при 300°. В технике способ дегидрогенизации циклич. соединений применяется для ароматизации нефти.т.е. для получения бензольных углеводородов путем отнятия водорода от нафтенов.
Водород как продукт разложения органических соединений получается иногда в больших размерах; каталитич. процессы имеют и здесь технич. значение. Carboneum-Gesellschaft получает водород разложением ацетилена под действием электрич. искровых разрядов, причем одновременно получается хорошая ламповая копоть. BASF получает водород пропусканием паров масла над Ктр., состоящим из MgO с 5% Ni, при 800— 1 000°. Ринклер и Вольтер впрыскивают в нагретые генераторы смолу или масло и получают водород и уголь; вдувая затем пары воды, они получают из отложившегося кокса и воды водяной газ.
Дегидрогенизация спи ртов применяется в промышленности для получения альдегидов по реакции:
C,Hs-OH=CHaCHO + H2.
Первое количествен, исследование этой реакции принадлежит Ипатьеву, к-рый нашел, что при пропускании паров а через трубку при f° ;? 700° идет двоякое разложение: 1) дегидратация и 2) дегидрогенизация. сна · сн.он
+ н2о
~*СН3 · сно + и5
Выход альдегида—не выше 30%, т. к. часть его разлагается по ур-ию:
сна-сно=оо + сн,.
В присутствии Ктр. (лучшими являются Zn, Fe и Си) темп-pa реакции снижается, и при 200—500° наблюдается значительный сдвиг равновесия в сторону образования альдегида, причем этиленовое разложение можно свести к нулю. С Zn можно получать 60— 75% альдегида, с Fe и Си—до 50%. Вторичные ы, при дегидрогенизации по Ипатьеву, дают кетоны с аналогичными выходами; третичные ы разлагаются при этих условиях на воду и непредельные углеводороды. Все сказанное относится также и к ароматич. ам. Путем пропускания метилового и этилового ов над различными Ктр. получают в заводских масштабах формальдегид и уксусный альдегид (Кот д Όρ, Бургундия,Шампань,Ганновер и т. д.). При применении тех же Ктр. под давлением характер распада ов меняется и, по терминологии Ипатьева, наступает «парафиновое» разложение, то есть получаются предельные углеводороды с выходами в 40—70%.
4) Гидратация. Каталитич. присоединение воды к непредельным соединениям этиленового и ацетиленового ряда идет по следующим ур-иям:
СН2: СЫ2 + Н20=СНзСЫ.ОН;
СН : СН + Н20=СНз ·
о н
Теоретически по приведенным схемам можно ожидать получения ов и альдегидов. Получение винного а из этилена и из ацетилена в настоящее время производится в заводском масштабе. В Англии из этилена, доставляемого газами коксовых печей, пропусканием его через серную к-ту, содержащую 5% СН20 и нек-рое количество ртутной соли, получают до 50 млн. галлонов этанола в год. В основе получения этанола из ацетилена лежит реакция Кучерова, к-рый наблюдал, что при пропускании ацетилена в растворы солей ртути образуется уксусный альдегид. Пользуясь этой реакцией, Нейман, Шнейдер и др. осуществили заводское получение этанола из карбида кальция: ацетилен пропускают через башни или ванны с H2S04, содержащей 0,5—1,0% HgS04. Альдегид получается с выходами до 90%, после чего его гидрируют над Ni, по Сабатье, в этанол. В качестве Ктр. применимы также Н2Мо04, Н3Р04 и др.
5) Дегидратация. В качестве водоотщепляющих Ктр. применяются самые разнообразные вещества: минеральные к-ты, их соли и ангидриды, хлориды металлов, окиси, органич. кислоты и их ангидриды, а также некоторые металлы. Дегидратация органич. соединений ведет к образованию непредельных углеводородов, эфиров и полимерных продуктов. В промышленности имеют зна чение глави. образом реакции образования олефинов и эфиров. Реакции дегидратации голен под влиянием Ктр. открыты Ипатьевым. Для получения олефинов из ов применялась ранее цинковая пыль, дающая выход до 50%. Ипатьев нашел, что куски графита с глиной и примесью железа являются хорошим Ктр., который при 500—600° превращает этиловый в этилен почти нацело, но лучшим Ктр. для этого процесса является глинозем, А1203, к-рый дает выходы этилена в 98% без примеси альдегида. Аналогично распадаются все жирные первичные одноатомные ы. Ароматич.спир-ты дают продукты глубокого уплотнения углеводородного характера; фенолы при действии А1203 дают сперва алюминаты, которые при повышенной 1° разлагаются с образованием эфиров:
2C„Ht · 0Н->2С1Н,5 0-С„Н5 + н,о.
Лучшие выходы аромат, эфиров достигаются с Th02; этот Ктр. применяют в заводских синтезах. Терпеновые н алициклнч. ы при дегидратации дают олефиновые углеводороды. Гликоли действием дегидратирующих Ктр. под давлением превращаются в окиси, которые затем изомеризуются в альдегиды или кетоны:
сн.он сн2ч си,
! }0 -> I
СН2ОН СН сно
Циклогексанол дает циклогексен, борнеол—камфен, ментол — смесь ментенов. Вторичные ы, равно как и третичные, дают лишь этиленовые углеводороды. Механизм дегидратации металлами и оки объяснить трудно; нек-рые авторы полагают, что Ктр. в условиях реакции имеет тенденцию к соединению с элементами воды. По этому представлению дегидратация ов идет в две фазы: сперва образуется эфир, к-рый далее теряет молекулу воды, превращаясь в олефин:
2 С2Н5011 ^ (C2Hs)20 + Н20;
(C2Hs)aO -> 2С,Н, + Н,0.
В атмосфере водорода под давлением при 400° расщепляется на эфир и воду; при 490° начинает образовываться этилен. Вторичные ы разлагаются быстрее, третичные—эфира не дают. Сабатье и Майль, применяя Th02, получали со ом комплексы вида ThO(OR)3, от которых действием NH3, НС1 и H8S можно переходить к аминам, галоидопроизводным и меркаптанам. Выходы эфиров с ТЮ2 достигают 50%. При пропускании смесей паров фенолов со ами при 390—420° над Th02 получаются смешан, жирно-ароматич. эфиры. Обычные способы получения сложных эфиров из к-т со ами также основаны на каталитич. ускорении реакции этерификации, что достигается действием сильных к-т (гомогенный К.) или пропусканием паров а и кислоты через нагретую трубку. Здесь Ктр. служат некоторые металлич. окислы, способные давать нестойкие соединения со ами; на 1 молекулу кислоты берется до 12 молекул а; выходы достигают 80—90%. На описанных реакциях базируется современное производство простых и сложных эфиров.
6) Пол и меризация и конденс а-ц и я. Среди многочисленных реакций полимеризации, проводимых при помощи Ктр., технич. значение имеет альдольная конденсация двух частиц уксусного альдегида в присутствии НС1 или ZnCl2:
,.о о сн, с< сн, с г -»· сн» си (он) · сн, · сс н чн чн
При нагревании ацетальдегида до 100° со стружками Ζιι получается кротоновый альдегид, СН3-СН:СН-СНО. Бензальдегид при нагревании со овым раствором KCN образует бензоин:
С,Н3 · СОН + СОН · Cell,-» С«П„ СН (ОН) · со С8Н5. Совершенно аналогично получается анизоин из анизилальдегида, кумипоин из кумино-вого альдегида и фуроин из фурфурола. Вероятно, большое техническ. значение приобретет реакция, впервые открытая Бутлеровым,—полимеризация формальдегида во фруктозу под каталитич. действием извести: в СН,0 -> СН,ОН (СНОП), со · сн2он.
Если пропускать пары метилового а в смеси с СО над Th02, ТЮ2, Ζη, Μη при 175—260°, то получается уксусная кислота. Очень интересны патенты Дрейфуса, к-рый получает уксусную к-ту из метана и С02 в присутствии солей Сои Ni. По другому способу того же автора, СО барботируют через теплый метанол, и парообразную смесь пропускают под давлением при ί° 300—350° через зерненый Ктр. (Sn02, ZnO, А1203 или Сг203); получается уксусная к-та, а при избытке метанола—ее метиловый эфир.
Реакции конденсации имеют большое техническ. значение в производстве и с к уест в е и и ы х смол, получаемых конденсацией формальдегида с фенолами, кровью, фурфуролом, кумароном, инденом и друг, (смотрите Смолы искусственные). Применение разнообразных Ктр., которые в данном случае называют модификатора м и, позволяет получать смолы различной окраски, мягкости, плавкости и твердости.
7) Галоидирование. Введение галоида в органич. соединения в присутствии Ктр. имеет технич. значение при хлорировании метана. При пропускании смеси СН4и С12 над СиС12, распределенной на пемзе, при 435°, получается смесь, состоящая из 5,4 ч. СС14, 4 ч. СНС13, 22 ч. СН2С12 и 0,6 ч. СН3С1. Хлорирование этана и высших парафинов может производиться с J, Р, Sb, Мо, А1С13 и FeCl3. Хлорированием в присутствии FeCl3, Fe203, FeC03, FeS04 легко переводится в хлорал. При бромиро-нании ароматич. углеводородов, в качестве Ктр. можно брать А1Вг3, FeBr3, Fe и друг. Присоединение галоидоводородных кислот к непредельным соединениям без Ктр. идет плохо даже при 150—200°; лучшими Ктр. для этой цели являются BiCl3 и BiBr3, которые позволяют вести реакцию даже при обык-
. иовенной {°, а при 120—150° проводят ее очень быстро. Бибо (Wibaut) получал хлористый этил из светильного газа (в к-ром всего лишь 1—2% этилена), действуя на него НС1 в присутствии ВЮ13.
8) Отрицательны и К. Отрицательные Ктр. имеют практич. применение лишь в целях «стабилизации» продуктов, когда необходимо предотвратить или замедлить химии. изменение вещества. Вода, которая в малых количествах играет роль положительного Ктр. для большинства химич. реакций, в больших количествах нередко оказывает отрицательное действие, замедляя, например разложение аммиака, а также и обратный процесс синтеза его из элементов. Многие соединения изменяются вследствие авто-окисления (смотрите); сюда относится, например, окисление альдегидов при их хранении. Предотвратить этот процесс можно прибавлением небольших количеств фенола, к-рый каталитически тормозит автоокисление. Тиомоче-вина, гидрохинон, фенол и другие вещества предохраняют многие тела от полимеризации, разложения или окисления: такие отрицательные Ктр. носят название ста б и-л и з а т о р о в; те из них, которые специфически замедляют окислительные процессы, называются также антиоксигснами (смотрите). 30%-ная перекись водорода (пергидрол) разлагается довольно быстро, но прибавлении барбитуровой к-ты, фенацетина или некоторых производных мочевины значительно замедляет процесс, так что продукт выдерживает перевозку и хранение. Хлороформ предохраняют от разложения прибавлением а, выцветающие краски стабилизируют фенолом. Защита металлич. поверхностен от окисления и коррозии достигается применением СгС13, к-рый защищает железо от действия воздуха и слабых к-т; таким же образом действуют хроматы и бихроматы. Соли железа предохраняют медь от коррозии.
Большое значение имеют отрицательные; Ктр. в области двигателей внутреннего сгорания, где в настоящее время широко применяются т. н. антиы (смотрите), прибавляемые в небольших количествах к жидкому горючему. В качестве антиов применяются: диэтилселенид, (C2H5)2Se, ди-этилтеллурид, (С2Н5)2Те, тетраэтилсвинец, (С2Н5)4РЬ и другие вещества.
Лит.: Ипатьев В., Роль окислов в явлении катализа, «Ж», 1907,- е г о ;к е, Каталитические реакции при высоких температурах и давлениях (докт. диссерт.), СПБ, 1907; Учение о катализе, сборник «Новые идеи в химии», в, Л., 1924; Д ж о б л и п г 1·:., Катализ и его применение в технике, М.—Л., 1925; Сыр к ин Я. К. Катализ, «ЖХИ», 192В, т. 3, стр. 1034, 1 110, 1197; Ч е л и н ц е в В. В., Кон-тактно-каталитич. процессы в области органических соединений, Л., 1927; Р а б и н о в и ч А. И., О механизме гетерогенных реакции, «Сообщения о научно-технич. работах в Республике», вып. 22, Л., 1927; Woker G., Die Katalvse, Die chemische Analyse, hrsg. v. В. M. Margosches, B. 11—12, Stuttgart, 1910, B. 21—22, Stuttgart, 1915; Henderson (>·, Catalysis in Industrial Chemistry, L·., 1921; Mail he A., I2tat actuel de la catalyse. Les actuality de chimie contemporaine, publ. par A. Haller, s6rle 1, Paris, 1922; Falk K. O., Catalytic Action, N. Y., 1922; Pascal P., Syntheses et catalyses industrielles, P., 1925; It i d с a 1 E. and Taylor H., Catalysis in Theory and Practice, I,., 1926; Mittaseh A., Bemerkungen z. Katalyse, «B», 1920, .!g. 59, p. 13; Structure et activite eliimiques. Rapports et discussions, Institut International de Chimie Solvay, Paris, 1926; Sabatier P., Die Katalyse in d. organischen Chemie, 2 Aufl., Lpz., 1927; Eiieal E., Catalytic Action, «Chemical Review», Baltimore, 1928, v. 5, p. 67; Bronsted I„ Acid and Basic Catalysis, ibid.,p. 231—338; Homogeneous Catalysis. Discussion, «Trans, of the Faraday Soc.», L., 1928, v. 24, p. 545— 696; Frankenburger W., Neuere Forschungen auf d. Gebiete d. heterogenen Katalyse, «Z. ang. Ch.», 1928, Jg. 41, p. 523, 561; Green B. ·Τ·, Industrial Catalysis, L·., 1928: Η i I d i t c h T. Catalytic Processes in Applied Chemistry, L., 1929. В. Ипатьез.
03
X
X
X
X
s *
g§
ЕН ей ей Я СО ей Я Я R со ей О Н
5 к
* О 03 О
ш°
а" а а ь В,
* н
X £*
1г я к
2 t
3 a
я я
£ я
« я о
* о й о о f-§»
=г
О — г-н. >_|
CL ? 2 b*
С Ϊ ^ ^
* ГС я
X
X ш
о
03
X
! Ни • аз η a о аз аз л ω §« и a
R g аз η Ен аз 5
s
д
cs
I-
CS
X
►£>&
§1»
gp
« в
a <υ И 2
w S
о 3
Ь Н
2 к к 3
я я ЮМ аз ад,
Ен Q3
о а н я
>* аз а я
s
| ей | Д ей | Я
сй |
1 | а и | is *
S .25 § |
№
Я 03 |
|||||
| И
О Еч СЗ а о |
О
’о оз Οχ М ей |
и
s 1 |
а
•о я НД я дд |
Я
03 J Еч ей а |
б ей | о я CQ О оз *Н и д н
С Сй ю я |
Ен я д | Ен
03 я |
3 О S §§§
“Й - оз7 р |
йЗ
Я 03 СЙ 03 |
ья±!
§» Ен - S.SP F3 g |
| E-ι
Я < |
;о оз £ л о | сй л
W |
ей | S | ЯЙ
СЛ |
Ьч Д
03 ей |
об | 5 | Сн ф —д Д-р я я р Д | я
03 |
ia
03 |
| а «я | О | ей | я | J=<3 ей | д | Сй | |||||
| Д | сл | СЙ | ей |
s с-
к а ьн и о “ я н а ей я Я а и · н з *г ° 3 S
-SB
Й Я р
S я 3
Н ** ей СЗ 03 Ен Я Я ей
gag
Сц
% О, Я Ен. я Сй ;
£ 5 § 2 Й
Я §
Я ей
Я
>>
S
я о
CQ О
. 3
«2 ιτπ <ζ К о я а ~ Я о Αυίί
•51
“ Щ R
„ о я
03 СЗ £-*
со
-X
2·^
►HQ
°о
До
J, ji, _г ьо ы» я &(<<! Д
ей СЗ. .„ а Я О Я ес
s
5 к“
• - я .>7
sW°
О пД-Л О
I I
ей μα —1 СО
я • •Д .·,
- 3 о я ^ со я асоа
СР
о
-<д уО
д-91 1
.д о О tn ’£ UtN
и?."« go® sM“
О
*«
Or
- н I I
ДО О СЗ о“вЗ_
за-„s„-
" J5 Л 3 С-ι
д я сй ^ s -
ни Я ft а ей о .ОО^
о
Яг
I -:чо «о
ιΟ а
г - *_г д я я 3 .Я
iMsHgN
ooo§ Рώ
я я я я R Д
о t<
с д
| S | г и 1000* | тН тЧ | ю со со | о
О 1<3 |
*о | |
| с | 1 03 о со | л | л | 1 J.
(М |
1о А А о | V I |
° и g s
о о со о сантиметров н<
о о
<М О (М СМ
о о о со
<М Ή
WfcRtSfci
^j3"l
i£Qf^
„. - a t© -ι-Я оо ^ »03 Ug«s
rrwi^ Ч .»· ·«*
1+ .1§о
»Ь^ О.ИО -•н Я Я О) О
»7^2 2 ЬД
Z+ §hS
ф-я
Ь< Д Осй И +
ьсоз С <!д а
-+ я
^3 о" o°g “о
._га 1 о я о
2
о
2
о ; я > я f
-, О ί
^ 2
о
2
5 з g 2-я
SS
‘52 + «
s о а г
>Ю RpJH.
* аярч^я
СЙ Я Д
s Bg -о
<3»
-S
О
я S о Й о я х Л Я
ΟΌ
9 Рн я ^ °
оа§5л
^ 5 ° -н я § .«irjz; Ч тзО
8«^2 > ® нГ ) g fL «
ЙЙ
Й Й
о"
О
о аз
fr
О I
R
зд
о
Й
a R
н >>
к н ей Я
.а ела я, я
-н О
. Йр“
г“*
.я аз а а >< я й б я^
РД н Д
оЛ со <м ей j·
КкГ
СЙЙ
а~
ig
03 о со аз О
Ен М
X
Я«з я а Я я £н аз аз Я ИЯ
« и а оз
So
§g
Ig
я я о %
2*^ я а а о и я Рч оз я;
Я м
° Я
я «
1>Н
2 s с
03
а о Ен
Н Се,ей н И и
Я Η Е
Н 03 с
Я И ;
К Я S-Я Н
Я В со о
| 03 | g я | 03 |
| я | за | я |
| о | а | о |
| н | в * | н |
03 г.1 „
Я E Е
до.с
ей н Ow, °?Я ь аа ϊ gat и a i я а < а и?
и
Ен ей О Я
й Й w Ч« VS_
о ?н м g я х» а
Я О ® « S Э lS К rt S мЛ 2 S В t О J и *5? 3*я l^js, Й л
п а Д оз м ^>8 2 ал н И аз со Я >0<-|-
isafs
я а з ы йКййй СН Я о Я
03 Ен ЕН н
О Яг Я Я1
PQ я И
я Я
s5hf
3 tol
йм
-Я)
Я· Я Ен Я 3 Я м Ь
И я я Я
я а« ж
Я 03 я I СЗ Я Н ет
Stgffi
«Ρ’α.
«Н о т ей:;
- 03 И
s3 я
:Я
3
Я н я а я a 2 Я i e- оз ао рй оа, а я а я о f я 1 о a U я i
я н at-
« ,. Р ей
В а и н я »».
<1 и ‘
в
О
И
Я
и
<
Иг-sO
SSgK
S“3S
ей 03
;*2 2 и
< о а я
> g о «
!
’ Р i и !^s9 ! я а А) о>с?
! Λ
t
О
О
H ft *
5 о ^
", Η irf
5 л к ·$ со о. а ·
° л s
с“ г“
г? S? 5
33*
а « л н. а л
- 05 £
л а а о н
НОЙ
о а о о а «·
!-f Й w
о С
« 2 я I
О 05
3 н а гй
« О)
R ^ ю а
| н
45 |
<о и
05 |
Hurler | о
С о5 |
о,
?3 ° бо а со _о^ |
| S | 1 Р 1 со | О | g С ω | |
| С. | с | в | tr‘· | -§*:? |
| о н о | CQ | о а | а с | о|« |
| < | о
•л |
Нч | б | к · Рг ·
h |
"|S t®
Л С, и ir из
Р=£5
* *Яий
„-§«» ·=J «;>,s
05 - Й 05 ^
й К Ojh И
£|=Уз2
|£6»й а _ сэ
"Г r/~^ о оО йс н a t-(
<0 Ξ>
а о
О
В
«м«"§
4*11
?-в“1§
ай.ий
Н «нн К® feP ® ^ίϋο 2
2 i
ISI I I
I I I
о о
£ о
3 И я 2
О 2
а Е и £ О н
Яд“
Orii
«л
н3
3 £
м О
Sg
й с он
-iXOSO
2-й о о
gSpS
3 4 о < -> а о « go £ Ipaa
^ * Οϊ-
I gill!
я >*§ >* £ а g w 5 w о 6
й о
°g
3
| со | w | |||||||||||||
| /л | т | л | V/V/V | V | V | |||||||||
| -470 | о о тЯ | о о
00 |
й оо | о ю
tf |
-360 | ч О О
о о со ю |
о о
СО 1 |
ООО
о о о ю со тЯ 1> IIIм |
350 | о ю со
1 |
500 | -650 | -350 | о р т |
| о
*о |
i
о |
i
о со |
о о
OJ |
о о
05 |
§
OJ |
500-
450- |
о о ю | О О о (
о ю со > 05 СО |
5 | о о со | ί | о о
»о |
200- | 250- |
о б
.„ н 3 2 ^ «
ей
!?В Рн с р^«
О «СЛ X*×ЬС
а ^
.й е, ^а +
ЙО
.Со ρ,Ο
^ "Ь
о о:§ ~
о 2оо
® Ос”"!
I ·*>
о5 н й 3
а
.1?« а а
О и о о й° «
Й км »
|SS®-*§+£ ·“!=-Bbgs-f st ,й о ^
о: й ооооо
j£’ о Н 2
Q^SoS
_ „se
SO
.„otiS
°Sgp I
« И p-
i-Suo
о
«o°
й «—Λ *“ p ;ϋΟ .Д(й
^ioso°±f
slgbl I
:C
a
or
S°.o
9.0
|9,fc HS-s 60 «9.
<!θ c
О
ρ-’Ο о CO
cT £
cgO r.Pn О g
9 6
t
о
О о о в
rfffi vfP
га i г -ч
«·ς «о
С Ew
с. О _ + оо ίο «t »9. о I с ίο о с—
« У, (М
а + а
«О
о1^ Я ~
о О
55
а со
°Я
gt
‘Н
ЯЛ» §
J3
а о 3 r ?
С.И (ί·!
О
| Л | Ч- н | 1
<15 |
1
О |
|
| а | о <0 | X | н | &oBS |
| о а | о л | f | а | ^s+g, |
| t | ogt | б | з | н“С8 § .ai |
| Q | « Н *> | ю | R | о К И |
| Ю
о S |
н шЮ «
О а >о |
tt? а + | а | я о н t, оЕв0: |
| + г s *«*
а ·+< |
ζ©
η. 2 -i- |
^ J м £ Н £ | 3h*o
к |
* £^« + ! |
«О
о£
. 05 О £
a t S* в<Я
ЙмО °-н
Н £ ГС >0:
05 «О
нОй о а о“ 2
б г° * л *
t
О
г·* г а С? н о о
gs
si
0.Н
ц&
:В 1“ 3 0«
§ + § S § a r
Л О о Сн ЯСНО rt g
w OjV
о а
К С
-ч и-
О >> г=В
S Ь-
о а н а и о
- с
-а Я н Рму г1 Hf и IS Е
at1*%*
Г MS3 а ы ’g· о а и о 12 а 05 Й
Й о а а
Н и-Н
Я- а о.
(Продолжение.)
А
E
S’ я
Н м
Ο
I I
α> Η,
в В s
I I
I I
8 B:
ί Э
° И
В ж я В
| В | о | о | о | ||||||||
| о со
1 |
В В В
о * |
—220 | §
I |
аз
В |
о
§ 1 |
о о
Т |
а | о о ю | тР
т |
со
7 |
С1
7 |
| о | 2 А | 130 | о со | В
0) |
250 | 250- | аз | S | 900 | о о ю | о о
о |
| О | В | гЧ | -Ч |
о
A
a
о
a
a
a
a
t=c
a
A
a
Ph
О
со
I I
о a я д
sS
«* §
t ё
a M’S a о a в я я
sgs
а§й
a о <d о m о К ж о о а о А £* А
«В н в~
^ Л
rr в 2
л а А <и
<1е о
W
m
α>
«5
_ В
а м я о|
в а а> 2 Ζ в
Эй t g а м
<В
в в а в в а? аз В В
В в ч«
аз аз о о
О О
о о к СО
a «в
S
в и
ОО
Ό®
рп
»о св -О
«Рч
>0<
в в
a
в
. я в й
Я’Ё
Н ® В Я
в яз н :В ° о 2
S-ю S
«СОЙ о 5 5 АО S
О А Я
«в
0 о В
1 и!
!§3s
§2§к й!®
А
со
^ a
® в я о
Ви« О в
5 я «
В В —
®аЗ ь,
2 ^ й
ί-Ал о +
ОО
в£
я
°53
««я
В + 03
“st
я *»о
ECO
£§#
я! «н
©В f
θ§ο
&“я
gg°
e,sj
к 2°
§ Э®
в£ +
8 ь
Еч СО
2 *
в
:В «° » »? Я © + о «jz
°rto
^ E-· В
л OZ
<М
. t
55 +
^ aJQ
ф ΰτ#
a f н 1М
§Й 18+ go £эт„ Я+О
a «R
аз а?о со
0- 4 +
1- S
В Р
я я „
О>&о
Я 0J
gg=i
Зой
В _
«Й
S»
ёв
>.<3?
к t
я «
ΨΖ
я „ до 5 ^ к?
СЭ о Я