 > Техника, страница 52 > Кинетика химическая
> Техника, страница 52 > Кинетика химическая
Кинетика химическая
Кинетика химическая, отрасльтео-ретич. химии, изучающая скорости химии. реакций. В основе опытной методики этого изучения лежит определение концентрации прореагировавшего либо появляющегося вещества в функциональной зависимости от времени. Различают кинетику о б-ратимых и необратимых реакций. К необратимым относятся те реакции, которые практически протекают лишь в одном направлении; это имеет место в случаях, когда продукты, которые образовались в результате химии. процесса, удаляются из сферы реакции и тем самым лишаются возможности обратного взаимодействия, или когда обратная реакция протекает лишь с незначительной скоростью. В случае обратимых реакций продукты вступают между собой во взаимодействие, давая исходные вещества; при
этом результирующая скорость определяется как разность скоростей прямой и обратной реакции. Далее, различают кинетику гомогенных реакций, протекающих в одной фазе (например когда все участвующие в реакции вещества газообразны), и гетерогенных, протекающих в разных фазах. К последним относятся реакции, сопровождающиеся образованием газообразных продуктов из твердых, переходом твердых веществ в раствор и тому подобное. Строго гомогенных реакций известно весьма мало; детальное исследование часто обнаруживает, что т. н. гомогенные реакции протекают на стенках сосуда и что их течение зависит от материала сосуда, а также от примесей твердых веществ, на поверхности которых может происходить адсорбция или, в общем случае, такое изменение свойств, которое делает молекулы активными, то есть реакциеспособными (смотрите Катализ).
Скорость реакции зависит от числа столкновений соответствующих молекул, поскольку химич. силы проявляются лишь на расстояниях порядка молекулярных размеров (10~8 см). В связи с этим основная закономерность К. х. может быть выражена так: скорость реакции, то есть убыль первоначального (или прибыль конечного) вещества во времени (или, иначе, производная концентрации по времени) пропорциональна числу столкновений наличных молекул. Согласно кинетич. теории, число столкновений п различного рода молекул пропорционально общему числу молекул в п-ной степени. Если реакция протекает по ур-ию:
пА + mB-*D +.
и если к моменту времени f концентрации молекул А, В и D соответственно равны С, С г и С3, то скорость реакции v выражается ур-ием:
.Ур-ие (1) показывает, что скорость реакции с течением времени уменьшается по мере уменьшения числа молекул А к В. Постоянной величиной, не зависящей от концентрации,является fej—к онстанта скорости реакции. Только изменение энергетич. условий, например повышение 1°, влияет на величину fcj.
В зависимости от того, сколько молекул должно столкнуться для элементарного акта взаимодействия, различают реакции 2-го,
3-го и т. д. порядков. Известны однако случаи, когда нельзя сделать непосредственного вывода о необходимости столкновений для осуществления реакции и когда элементарный акт реакции заключается как бы в самопроизвольном распаде молекулы. Классическим примером такого процесса является распад N205, причем скорость реакции пропорциональна наличной концентрации данного вещества в первой степени и в значительных пределах не зависит от примесей посторонних газов. Если начальная концентрация равна С, а ко времени ί прореагировало количество вещества ж, то
b-ЫС- ж), (2)
или ж=С (1 - е-*1). (3)
В случае, когда для реакции необходимо п столкновений, уравнение (2) приобретает вид:
<£=кп(С-хТ (4)
И
ж=С 11 — [1 + (и — 1) кпСп-1 ί]Γ-”|. (5)
Наблюдая течение реакции во времени, находят путем подбора такое значение для п, при к-ром концентрация, как ф-ия времени, выражается ур-ием вида (5) при постоянном к. Так. обр. находят порядок химич. реакции. Опытные исследования показывают, что реакции высоких порядков весьма редки. Это находит себе объяснение в том, что одновременные столкновения большого числа молекул мало вероятны и происходят гораздо реже, чем парные или тройные соударения. Много реакций протекает по ур-иям кинетики 1-го и 2-го порядков.
Исследование скорости реакций позволяет выяснить их механизм. Обычное стехиометрии. ур-ие реакции означает только баланс начальных и конечных веществ, составленный так, чтобы зная—чтб реагирует и что получается, подобрать такие коэфициенты, при которых выполняется закон сохранения материи. Однако нет веских оснований для предположения,что реакция протекает именно по написанному ур-ию. Определяя кинетически порядок реакции, исследуя влияние избытка разных компонентов ее, выясняют механизм реакции и характер элементарных актов, в результате которых появляются новые молекулы. Весьма часто сложные реакции осуществляются через образование промежуточных веществ, обладающих небольшой длительностью существования и превращающихся при помощи цепи промежуточных реакций в конечные продукты. Каждая промежуточная реакция идет со своей специ-фич. скоростью. Измеряемая общая скорость реакции определяется течением наиболее медленной промежуточной.
К. х. в растворах осложняется ролью растворителя, влияющего на константу скорости. В этой области известно весьма мало систематических исследований. Для типичного случая образования иодист. тетраэтил-аммония, N(C2H6)4J, найдено возрастание константы скорости с увеличением диэлек-трич. постоянной растворителя. Последнее свойство находится в связи с ионизирующей способностью растворителя. Допущение, что каждое столкновение соответствующих молекул ведет к реакции, требует, чтобы последняя протекала с очень большой скоростью. Это оправдывается для ионных реакций. В большинстве случаев однако лишь незначительная часть столкновений эффективна. Исследование влияния t° на скорость реакций подтверждает указанную точку зрения. Небольшое повышение t°, примерно на 10°, влечет за собой увеличение константы скорости в 2—3 раза, тогда как число столкновений при этом растет весьма незначительно (пропорционально |/Т). Теория приводит к следующему соотношению между числом столкновений F, обусловливающих реакцию (эффективных), и полным числом соударений Z:
Е
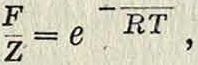
где R—газовая постоянная, равная 1,98 cal на з-моль, Т—абс. температура и Е—т. н. энергия активации, величина к-рой— порядка нескольких десятков тысяч cal иа з-моль. Из ур-ия (6) видно: а) что при малых значениях Т эффективные столкновения составляют лишь незначительную часть общего числа столкновений, б) что при небольшом повышении темп-ры F быстро растет. В основе воззрений, приводящих к уравнению (6), лежат допущения: а) что переход от начального вещества к конечному осуществляется через нек-рое промежуточное активное состояние и б) что молекула реагирует лишь в том случае, если она в данный момент находится на высоком уровне энергии, превышающем нормальное состояние. Активация может произойти при столкновении тех молекул, к-рые, согласно максвелловскому распределению скоростей, обладают значительной кинетической энергией движения. Теория активации, объясняющая К. х., дает возможность подойти к разъяснению явлений катализа. Большой температурныйкоэф. константы скорости характеризует резкую разницу между течением реакций на холоду и при нагревании. Экзотермич. реакции идут медленно при невысоких 1°, вследствие чего выделяющееся при процессе тепло успевает рассеяться. Повышение темп-ры ускоряет реакцию и следовательно вызывает более быстрое выделение энергии. При достижении некоторой определенной t° скорость реакции приобретает столь большое значение, что энергия не успевает рассеиваться вследствие теплопроводности и лучеиспускания и благодаря этому происходит дальнейшее самонагревание, еще большее возрастание скорости и вспышка. Так. образ. t° вспышки определяется, с одной стороны, кинетич. фактором (зависимостью константы скорости от ί°) и, с другой, совокупностью физ.условий (теплопроводности идр.).
Кинетика обратимых реакций учитывает обратное взаимодействие конечных продуктов, в результате чего вновь получаются исходные вещества и в общем итоге уменьшается скорость прямого процесса. Если процесс заключается в превращении А^Ви если начальная концентрация вещества А равна С, а количество, прореагировавшее за время t, равно ж, то скорость определяется ур-ием:
v=~=k1(C-x)-1c2x. (7)
Отличительной особенностью в данном случае является наличие двух констант скоростей: —прямой реакции и /с2—обратной.
Когда концентрации веществ А я В принимают такие значения, что скорости взаимно противоположных реакций становятся равными, наступает химическое равновесие (смотрите Действующих масс закон).
Кинетика гетерогенных реакций зависит: а) от скорости, с какой молекулы вещества достигают места, где происходит ре акция (реакционной зоны), б) от скорости самой реакции и в) от скорости, с какой образующиеся вещества удаляются из зоны реакции. В ряде случаев, как-то: при растворении твердых тел в к-тах или в инди-ферентных растворителях, скорость самого процесса весьма велика, вследствие чего около поверхности твердого вещества практически моментально образуется тонкий слой насыщенного раствора. Скорость процесса определяется в данном случае медленно протекающей диффузией. При максимальной концентрации насыщения С, поверхности О, коэф-те дифзфузии D и толщине пограничного слоя s—количество растворившегося вещества х ко времени t может быть найдено из уравнений:
| *)· | (8) |
| , D О · i. | |
| 1
1 т-Ч О II |
(9) |
Подстановка опытных данных дает для толщины диффузионного слоя s величину около 10_3 сантиметров и меньше. Присутствие посторонних адсорбированных веществ на поверхности твердого тела может сильно влиять на скорость. При гетероген. процессах, сопровождающихся образованием новой фазы (как например кристаллизация из пересыщенных растворов), фактором, определяющим течение процесса, является уже не диффузия: кинетика подобных процессов зависит от скорости образования зародышей и от быстроты их роста. Чем в большей степени переохлажден пересыщенный раствор, тем большую величину приобретает скорость образования зародышей, которая в определенной точке достигает своего максимума и при дальнейшем понижении ί° начинает замедляться. Скорость роста кристаллов измеряется приращением их линейных размеров в единицу времени.
Наиболее изучена кинетика следующих реакций: диссоциация молекул галоидов, распад метана, этана, ацетилена, ов, этилового эфира, формальдегида, уксусного альдегида, ацетона, муравьиной и уксусной кислот, окиси углерода, а, окислов азота, фосфористого и овистого водорода", хлористого сульфурила, никель-карбонила; синтез озона, воды, сероводорода, сернистого газа, серного ангидрида, аммиака, бромистого и йодистого водорода, хлористого и бромистого нитрозила, двуокиси азота; образование водяного газа, окисление углеводородов, бронирование их и некоторые другие реакции. Кинетика многих реакций изучена в связи с действием катализаторов.
Лит.: С ы Р к и н Я. К., О кинетике гомогенных реакций, Сообщения о научно-технич. работах в Республике, вып. 22—Первая конференция по физ.-химии. вопросам, Л., 1927; Н i nschelwood С. N., The Kinetics of Chemical Change in Gaseous Systems, Oxford, 1926 (нем. пер., дополпенный библиографией, Lpz., 1928); В о d e n s t e i η M., Chemische Kinetik, «Ergebn. d. exakten Naturwiss.»,B. 1,B., 1922, «Ztschr. f. Elektroch. u. angew. physikal. Chemie», Lpz., 1925, Jg. 31, p. 343; В e n г a t h A., Physikahsche Chemie, T. 2—Wissenschaftliche Forschungsberjchte, Naturwis-senschaftliche Reihe, hrsg. v. R. Liesegang, B. 14, Dresden—Lpz., 1925; см. ташке Катализ. Я. Сыркии.