 > Техника, страница 69 > Пассивирование
> Техника, страница 69 > Пассивирование
Пассивирование
Пассивирование электрохимическое, процесс, в результате которого металл делается неспособным к своим обычным реакциям и уподобляется благородным металлам. Напр. железо, будучи обработано конц. азотной кислотой, теряет способность растворяться в кислотах, выделять медь из раствора медного купороса, растворяться на аноде при электролизе и т. д. Способностью пассивироваться кроме железа обладают в большей или меньшей степени никель, кобальт, хром, свинец, марганец, алюминий, олово, ванадий, ниобий, .молибден, вольфрам, рутений, золото. П. металла часто наблюдается при электролизе; например если анодно поляризовать железо в разведенной серной к-те, то при небольших плотностях тока оно ведет себя нормально и переходит в раствор, давая сернокислое железо; если же путем повышения подводимого напряжения увеличивать плотность тока, то при достижении известной величины плотности тока, зависящей от природы раствора, в к-рый погружено железо, сила тока начинает внезапно падать и в некоторых случаях может стать даже равной нулю. Если однако приложенное напряжение достаточно для поддержания на анодной поверхности потенциала, необходимого для выделения кислорода, то прохождение тока разумеется не прекратится, но за его счет будет лишь выделяться кислород, а железо растворяться не будет. Следует отметить, что ставшее пассивным железо не будет растворяться и в том случае, если плотность тока будет вновь снижена до значения меньшего того, при котором пассивность наступила. Если ток прекратить, то в кислой среде пассивность обычно через некоторый промежуток времени прекращается, в нейтральной удерживается в течение значительно большего времени, а в щелочной восстановления активного состояния обыкновенно не наступает. Присутствие в растворе хлоридов значительно ускоряет процесс восстановления активности, наоборот, присутствие окислителей (хромовые соли, перекись водорода и проч.) обычно ускоряет процесс П. Низкая 1° благоприятно влияет на процесс И., в то время как при повышенной t° П. наступает при гораздо больших плотностях тока.
Процесс П. вызывает соответствующие изменения потенциала металла, погруженного в раствор, причем это изменение всегда таково, что потенциал металла становится более электроположительным (благородным); часто это ведет к тому, что пассивированный металл начинает переходить в раствор в виде ионов своей высшей степени окисления. Типичный пример этому имеет место у хрома. Нормально он должен растворяться, давая Сг” аналопгчно Fe~, однако если хромовый анод поляризовать в щелочном растворе, то только по достижении потенциала Eh— =0,62 V начинается растворение металла и притом в виде шестивалентных ионов CrVi. Последние немедленно реагируют со щелочью:
Crvi +8 ОН=Сг04"+ 4 Н80, в результате чего получается хромовокислая соль.Аналогично этому железо в щелочном растворе дает ионы Fe04", соответствующие солям железной кислоты (ферраты).
Наиболее общепризнанной теорией процесса П. металлов является в настоящее время теория, предложенная в свое время еще Фарадеем, объясняющая процесс П. возникновением на поверхности металла оксидной пленки. Основанием для такого воззрения являются следующие факты. 1) Пассивирование всегда связано с окислительным процессом либо с непосредственным воздействием окислителей, как кислород, озон, крепкая азотная к-та, соли хромовой кислоты, перекись водорода, либо с анодной поляризацией, что также связано с действием кислорода, выделяющегося на аноде, в то время как водород, восстановители, а также катодная поляризация металла способствуют обратному процессу·—переходу металлов в активное состояние. 2) Разница в условиях, при которых пассивируются различные металлы, а именно: железо и никель лучше всего сохраняют свою пассивность в присутствии щелочей, в то время как кислоты способствуют удержанию активного состояния или облегчают переход в активное состояние, если мы имеем дело уже с пассивированным металлом. Правда, в очень крепких растворах щелочей анодно поляризованное железо может оставаться активным, что объясняется большой растворимостью окиси железа в концентрированной щелочи; то же самое имеет место у кобальта, к-рый в разбавленной щелочи легко пассивируется, а в конц. растворе (8 N) легко растворяется под действием тока, давая растворимые к о б а л ь-титы. Металлы же, подобные вольфраму и молибдену, окислы которых имеют кислый характер, легко растворимы в щелочах и очень трудно в к-тах, ведут себя противоположно железу и никелю и пассивируются лучше всего в кислых растворах. 3) Возможность устранения пассивного состояния металла механическим путем (очистка метал лической щеткой), после чего металл становится снова активным.
Возражения против этой теории обычно сводятся к следующему. 1) Металлы при П. часто сохраняют свою поверхность блестящей, что заставляет сомпе-ваться в наличии яа их поверхности пленки окисла, тем более, что Мюллером и Кенигсбергером были произведены очень точные исследования онтических свойств пассивированных и непассивированных поверхностей металлов, не давшие никаких доказательств разницы в их оптич. свойствах. 2) Для того чтобы объяснить П. металлов азотной кислотой, надо допустить, что оксидпап пленка в пей нерастворима, что представляется маловероятным, ибо обычно окислы металлов хорошо растворимы в кислотах. В одной из работ последнего времени теория оксидной пленки пашла себе подтверждение, устраняющее в основном все возражения против нее. Фрейндлих, Пачеке и II ох ер показали, что если получить железо в вакууме без доступа кислорода (они получали железпое зеркало на стекле термин, разложением паров пента-карбонила железа в вакууме), то опо не пассивируется конц. азотной к-той, если же то же железо подвергнуть действию конц. азотной кислоты в присутствии воздуха, то П. наступает. Следовательно роль кислорода при процессе II. можно считать окончательно установленной; авторам удалось также доказать, что на поверхности пассивированного железа получается топкая пленка окисла, нерастворимого в азотной кислоте. Из других теорий пассивности можно указать на следующие. Леблан объясняет наступление пас-сивпости уменьшением скорости образования ионов, что происходит вследствие поглощения поверхностью металла кислорода, но последний не образует с металлом окисной пленки, а дает твердый раствор; по Тамману такого рода поглощение кислорода объясняется насыщением свободных валентностей атомов металла, лежащих на поверхности, кислородом, но характер кристаллич. решетки как для насыщенпых кислородом атомов металла, так и ненасыщенных, лежащих под ними, остается по Тамману одинаковым. Эту форму поверхностного поглощения кислорода Эванс назвал «двухмерной окисью». По теории «знач-ности» Крюгера и Финкельштейна у металлов, которые образуют ионы различной валентности, попы существуют уже в металле и находятся между собой в известном равновесии; так например, в случае железа имеет место равновесие между Fe··- и Ее^-ионами. При нарушении равновесия вследствие перехода в раствор двухвалентных ионов восстановление его происходит не моментально, что ведет к тому, что на поверхности металла, граничащей с раствором, остаются только Κβ···-ιΐ0Ηυ. Это вызывает соответствующее изменение потенциала металла и обусловливает более благородный характер пассивированного металла. Еще дальше такого рода взгляды развиты в теории Смитса, которым дана общая схема равновесия между атомами металла и ионами как в самом металле, так и в прилегающем к металлу слое раствора.
II. металла чрезвычайно благоприятствует сохранению металлов, т. к. оно парализует т. н. гальванокоррозию металлич. предметов (смотрите Коррозия). П. объясняется также устойчивость таких металлов, как алюминий и магний, против действия кислорода и воды: слой окисла, образующегося на поверхности металла, предохраняет его от дальнейшего окисления; так как окись алюминия и магния нерастворима в воде, то эти металлы в силу этого не разлагают воды с выделением водорода, как это они должны были бы делать по своему положению в ряду напряжений. Образованием оксидной пленки на поверхности алюминия объясняется также способность алюминиевого электрода пропускать ток только в одном направлении, что находит себе применение в устройстве электролитических выпрямителей (смотрите) переменного тока. Существование оксидной пленки дает возможность пользоваться некоторыми металлами в качестве нерастворимых электродов при электролизе (железо, никель при электролизе щелочных растворов, свинец при сернокислотных). Использование железных сосудов для транспортирования конц. серной к-ты, железных бал лонов для жидкого хлора и тому подобное. также основано на явлении П.
Лит.: И згарышев Н. А., Электрохимия и ее техническое применение, стр. 151—165, Л., 1929; его ж е, Исследование в области электродных процессов, М., 1914; К н с т я к о в с к и и В. А., Электрохимии. реакции и электродные потенциалы некоторых металлов, 1910; Электрохимии. свойства металлов, «Труды 2-й физико-химической конференции в Москве», Л. 1928; Ко erst er F., Elektrochemie WiTsseriger LOsungen, p. 4 15—446, Handbuch d. ange-wandten pliysikalischen Cbemie, hrsg. v. G. Bredig, В. 1, 4 Aufl., Lpz., 1923; Grub e, «Ztscbr. f. Elektro-chemie», Berlin, 1927, B. 33, p. 389; Haber und Goldschmidt, ibid., 1906, B. 12, p. 64; M ii 1-ler W. I. u. К 0 n i g s b e r g e r. Ibid., 1907, B. 13, p. 659, 1909, B. 15, p. 742; Le Blanc, ibid., 1900, B. 6, p. 472; T a m m a n, «Ztschr. f. anorganische und allgemeine Cbemie», Leipzig, 1919, B. 107, p. 104; Evans, «Transactions of the Faraday Society», L., 1922, v. 18, p. 1; Freundlich H., Patscheke und Z о c h e r, «Ztschr. f. phy-sikalische Chemie», Leipzig, 1917, B. 128, p. 321, B. 130, p. 289; Evans, «Journal of the Chemical Society», London, 1927, v. 1, p. 102; Gerdind u. К a r s t e n, «Ztschr. Г. Elektrochemie», Berlin, 1925, B. 31, p. 135; Foerster, ibid., 1927, B. 33, p. 406; Muller W. I., ibid., 1904, B. 10, p. 518, 1905, B. 11, p. 755, 823, 1924, B. 30, p. 401, 1927, B. 33, p. 401, 1928. B. 34, p. 840; Finkelstcin, «Ztschr. f. phv-sikalische Cbemie», Lpz., 1902, B. 39, p. 91; Smits A., Die Theorie der Allotropie u. ihre exp’erimentale Bestatigung, Lpz., 1921. С. Плетенвв.
П. адсорбционное, явление понижения скорости реакции на твердых поверхностях кристаллов и металлов, например понижение скорости их растворения в воде или к-тах, вызванное образованием на данной поверхности адсорбционного слоя полярных молекул поверхпостноактивных веществ. П. адсорбционное—явление отрицательного гетерогенного (контактного) катализа. В технике уже давно известно, что скорость растворения металлов в водных кислотах сильно понижается (в 10—100 раз) от прибавления к водной среде фенола, углеводов, сульфокислот, нафтеновых и жирных к-т и др., чем и пользуются практики, например для предохранения от потерь металла (при очистке поверхности листового железа от окислов) опусканием металла в кислую ванну (H,S04или НС1)—патент Фогеля, или например присадка а н т р, выработанная Степановым и Комовским в Химическом ин-те им. Карпова в Москве, и др.
П. адсорбционное вызывается веществами, поверхностноактивными по отношению к пассивируемой поверхности (адсорбируемыми ею). Механизм адсорбционного П. тот же, что и при понижении скорости кристал-лизации (си. Кристаллизация) поверхностно-активными веществами, растворенными во внешней среде—явлении, исследованном ранее Фрейндлихом и Марком. Полярные молекулы поверхностноактивных веществ, адсорбируясь на поверхности металла, покрывают ее при достаточной концентрации как бы кристаллич. мономолекулярной пленкой, препятствующей ионам металла переходить в раствор и замедляющей установление равновесия между металлом и раствором (смотрите Капиллярные явления, Поверхностное натпоюение, Полярность). С увеличением концентрации поверхностноактивного и а с-с и в а т о р а скорость растворения металла понижается, стремясь к минимальному пределу, соответствующему образованию насыщенного адсорбционного слоя наметаллич. поверхности. Для количественного исследо вания П. адсорбционного надо измерять кинетику растворения металла в чистой водной к-те (например в 10—20% H2S04, НС1) сначала в отсутствии (фигура 1, кривая а) пассива-тора, а затем в его присутствии при разных концентрациях (кривые Ь, с </, е) с сохране
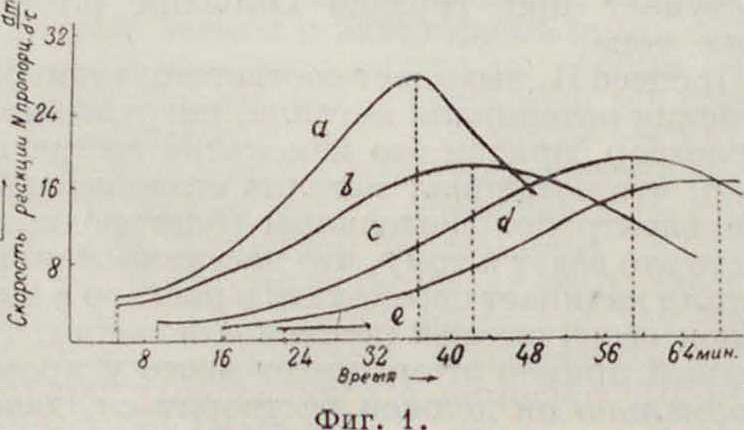
нием постоянства объёма раствора и при условии,что пассиватор не изменяет практич. состояния минеральной кислоты в растворе.
Для измерения скорости растворения металла или кристалла, например кальцита (СаС08), в к-тах удобно измерять число пузырьков газа (Н, или СОг), выделяющихся при постоянных условиях 1° и давления из остро обрезанного капиллярного кончика определенного диаметра в единицу времени (за 1 мин.). При этом N пропорционально скорости реакции где т масса вещества,
перешедшего в раствор в з, а т время в ск. Схема установки для измерения скоростей представлена на фигуре 2. Растворение про
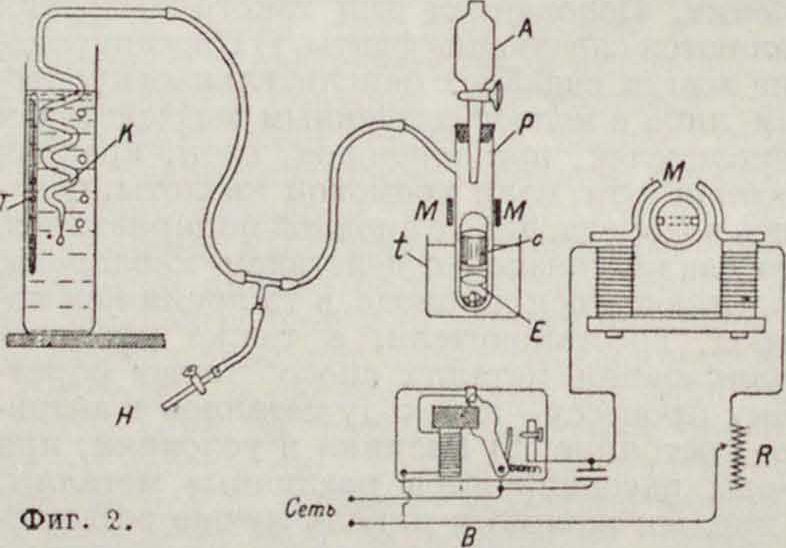
исходит в пробирке Р; капельная воронка А служит для добавления поверхностноактивного вещества, а кран II для сообщения прибора с атмосферой. Пробирка Р при измерениях обыкновенно помещается в водяной термостат t. Для перемешивания раствора в пробирке служит электромагнитная мешалка Е из согнутой толстой платиновой проволоки, заканчивающаяся внизу площадкой из платиновой сетки; на эту площадку помещается кристалл кальцита или к мешалке тонкой платиновой проволочкой привязывают полоски из листового металла. В верхней части мешалки помещается платинированный железный полый цилиндрик с (толщина стенок в 0,5 миллиметров). Мешалка приводится в действие сильным электромагнитом М, между полюсными наконечниками которого помещается пробирка. Постоянный ток, питающий электромагнит, прерывается посредством маятникового прерыва-
теля-реле В, позволяющего изменять число перемешиваний (колебаний мешалки) от 30 до 120 и более в мин. Кроме того перемешивание можно ослабить или усилить перемещением электромагнита М вдоль пробирки или изменением силы тока с помощью реостата II. Пузырьки газа при растворении вещества в пробирке Р выходят из кончика капилляра К, погруженного в ванну-термостат Т. Пузырьки газа должны выделяться в жидкости, насыщенной данным газом и практически его больше не растворяющей (наир, в крепком водном-растворе NaCl).
Прекрасными пассиваторами являются чистые фенолы, ароматические углеводороды (бензол, ) в тех ничтожных концентрациях, в которых они растворимы в водной
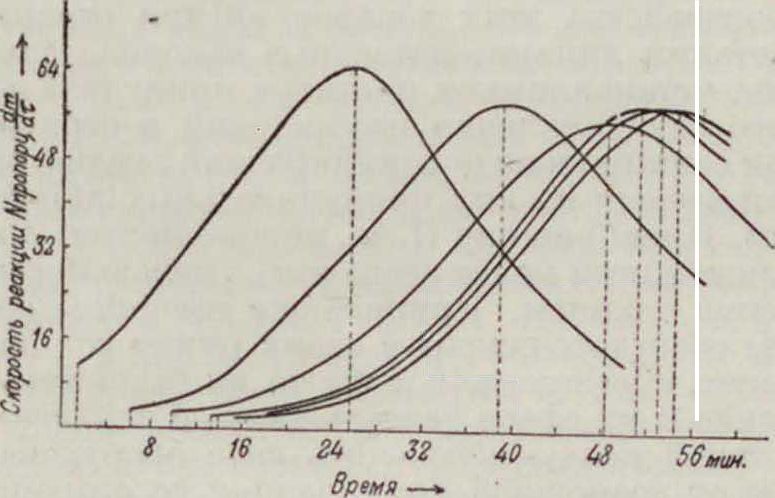
Фигура з. среде, ы, жирные кислоты, сульфокислоты, камфора и разнообразные смеси этих веществ (Ребипдер), могущие служить для изготовления новых рецептур присадок. На фигуре 1 видно, что вначале скорость растворения металла как в присутствии пасси-ватора (кривые Ь, с и d), так и без него (кривая а) обычно растет со временем, очевидно благодаря увеличению поверхности при разъедании (поверхностному диспергированию—разрыхлению). При этом пассп-ватор,уменьшая скорость растворения, уплощает всю кривую кинетики (кривая е). В дальнейшем же (при малом количестве к-ты) она быстрее расходуется в отсутствии пас-сиватора, что в таком случае влечет за собой пересечение кривых кинетики (фигура 3). И. И. Жуковым и его сотрудниками было показано, что поверхностиоактивные пасси-ваторы затрудняют установление нормального потенциала на границе «металл—раствор», чем и объясняется известное явление отравления, например водородного электрода, поверхностноактивными веществами.
Лит.: Ребипдер П. А., Ребипдер К. П., Пассивирующие (отрицательно-каталитические) действии адсорбционных слоев поверхностноактпвных веществ, «Журнал физии, химии», M., 1930, т. 1, вып. 2 (указана литература). П. Ребиндер.