 > Техника, страница 71 > Пирогенетические процессы
> Техника, страница 71 > Пирогенетические процессы
Пирогенетические процессы
Пирогенетические процессы, пирогенные процессы,пиролиз,пирогенизация, пирогенез, в широком смысле слова химич. превращения, происходящие под влиянием высокой темп-ры и заключающиеся в превращении исходного вещества в другие (без обязательного химич. воздействия посторонних реагентов). Физи-ко-химич. смысл этих превращений заключается в том, что при сообщении телу тепла происходит накопление энергии в молекулах, которые согласно закону распределения Максвелла приобретают способность к изомеризации или разрыву с отщеплением отдельных частей и превращаются в более простые молекулы, обычно более устойчивые при высоких темп-рах. Наибольшее технич. значение имеют П. п., связанные с органическими веществами естествен, происхождения. Вследствие сложности происходящих при этом химич. процессов до самого последнего времени многое являлось еще далеко не исследованным, и применение этих процессов основывалось гл. обр. на эмпирич. данных; только в самое последнее время создается более твердая научная база для понимания этих важнейших промышленных процессов.
Основными типами П. и. в технике являются: 1) Коксование (смотрите) каменных углей— термин, обработка их при весьма высоких темп-рах (900—1 250°), связанная с весьма глубокими изменениями исходного вещества и одновременно с образованием больших количеств наиболее простых молекул в виде газов. 2) Швелевание (смотрите)—низкотемпературная сухая перегонка каменных и бурых углей, горючих сланцев и других битуминозных пород. Характерной особенностью этих процессов является применение более низких темп-р (450—500°) с целью избежать глубокого распада углеводородных продуктов, являющихся смазочными материалами и жидким автомоторным топливом как замена наиболее ценного горючего—бензинов и керосинов нефт. происхождения. 3) Сухая перегонка дерева, см. Дерево, сухая перегонка.
4) Пирогенетич. разложение нефти и ее продуктов наряду с коксованием углей является одним из наиболее важных в технич. и хозяйственном отношении технологич. процессов. Пирогенетич. разложение нефтяных продуктов по своей целевой установке м. б. разделено на 2 основных группы: а) ароматизация, высокотемпературный процесс (600—700°), имеющий целью получение низших ароматич. углеводородов, гл. обр. бензола и а, и б) крекинг-процесс (смотрите), П. п., протекающий при более низких темп-рах (425—500°), ведущий к образованию автомоторного топлива: бензинов и керосинов.5)Деструктивная гидрогенизация, или крекинг в присутствии водорода, с формальной точки зрения можно не относить к П. п., так как участие .в процессе водорода в качестве химич. агента выделяет ее среди типичных П. п. Однако по характеру процесса деструктивную гидрогенизацию можно считать в значительной мере аналогичной обычному крекингу.
Химизм пирогенетических процессов. Основными процессами, имеющими место при нагревании органич. веществ до высоких ί°, являются: 1) термин, расщепление (дезагрегация); 2) полимеризация и уплотнение и 3) дегидрирование и гидрогенизация. Рассмотрим химизм в отношении отдельных классов углеводородов, заслуживающих с практической точки зрения наибольшего внимания.
1) Метановые углеводороды. Основной реакцией термич. превращения насыщенных алифатич. углеводородов является указанный еще Вертело I1] распад по схеме
Cm+nH2<m+n)+2 -> СтНгш+г + СпНгп с образованием молекул метанового углеводорода и олефина. Относительно места разрыва цепи и влияния темп-ры, давления и др. достаточно обоснованной теории еще нет. Экспериментальные наблюдения, касающиеся например влияния повышенного давления на характер разрыва цепи, скорее всего могут быть объяснены температурным режимом крекинга в жидкой фазе, когда продукты разложения не подвергаются столь глубокому распаду, как это имеет место при процессе в паровой фазе, всегда сопровождающемся явлениями значительного перегрева и потому обусловливающем более глубокое дробление молекул. Именно этими, б. объяснено значительное образование газов при П. п. под атмосферным давлением. Однако если нормальные углеводороды склонны при первичном расщеплении распадаться по приведенной выше схеме, то молекулы с разветвленными цепями могут отщеплять в зависимости от структуры скелета также и преимущественно короткие осколки цепей или даже водород. Поэтому ххоличеетво обра зующегося газа при технич. пирогенизации в значительной мере зависит от характера исходного материала. В зависимости от температуры и времени пребывания в реакционной зоне степень дезагрегации исходного продукта меняется и может достигнуть при достаточной темп-ре (выше 1 000°) полного распада на элементы. С увеличением молеку-лярного веса термическая стабильность метановых углеводородов понижается. В метановых углеводородах нормальной структуры происходит также отщепление водорода. При наличии же изостроения дегидрогенизация получает более важно© значение; так, при пиролизе изобутана до 50 % его превращается в изобутилен [2] по схеме:
(СНзЬСН СНа -> (СН3)2С : СНг + Н2.
2) Непредельные углеводороды жирного ряда. Содержащие этиленовые или ацетиленовые связи углеводородные молекулы распадаются аналогично метановым углеводородам, по схеме например: C(m+n)H2(m+n) СтН2пц2 + СпН2п-2, с образованием насыщенного углеводорода и еще более непредельного (например с двумя этиленовыми связями). Точно так же возможны реакции отщепления водорода; например этилен при 800° дает до 3,5% ацетилена. Имеют место при высоких темп-рах также реакции изомеризации: осколок нормальной цепи приобретает изостроение. Однако наиболее характерными реакциями олефинов, а также ацетиленовых углеводородов при П. п. являются полимеризация и уплотнен и е. Работы Доу, Ипатьева [3] и др. показали способность этилена и его гомологов давать циклич. соединения—нафтены, которые являются более устойчивыми при высоких темп-рах, по схеме:
сн2
НгС/ЧСНг 3 СН2 : СН2 -> I
Н2СЧУСН2
СН2
Кроме того при этом возможны реакции уплотнения олефинов с образованием более высокомолекулярных ненасыщенных угле-водородов; например амилен дает полимеры: ди-, три- и тетраамилены. Все эти реакции уплотнения хорошо протекают уже при 400— 450°, а в присутствии некоторых катализаторов, наир, фтористого бора, идут уже на холоду. Сувеличением молекулярноговеса, олефины становятся менее устойчивыми в отношении темп-ры и приобретают большую склонность к реакциям уплотнения. Особенную способность к образованию высших полимеров имеют диэтиленовые углеводороды: эритрен, изопрен и др. Продукты полимеризации этих углеводородов, полученные при низкой темп-ре в присутствии специальных катализаторов, дают искусственный каучук. Из числа многих предложенных методов получения диолефинов наибольшие перспективы имеют пути, лежащие через П. п. Так, для этого может быть использовано разложение при темп-рах 500—700° нефтяных продуктов [4] или термич. разложение ов [5] или конденсация при 350—450° этилена с ацетиленом [·]; последние м. б. получены также пирогенетически, и т. д.
*и
3) Кольчатые углеводороды ряда нафтенов. Близкие по своим химическим свойствам к метановым углеводородам насыщенные нафтены в условиях П. п. более устойчивы. Реакции расщепления идут прежде всего в сторону отрыва боковых цепей, причем легче всего отрываются наиболее длинные цепи, а наиболее устойчива метальная группа. По лиметиленовое кольцо способно также разрываться при высокой темп-ре с образованием ненасыщенного углеводорода с открытой цепью, напр:
СН2—СН2
сн2—сн2
СН3СН2СН:СН2
К реакциям уплотнения насыщенные циклические углеводороды, как и метановые, мало способны. Весьма своеобразны реакции изомеризации полиметиленовых углеводородов, заключающиеся в изменении числа членов кольца. Так, шестичленные кольца при повышенных темп-pax могут переходить в пятичленные [7] по схеме:
сн2 сн2
СНг^СНг СНа^СН-СНз
СНг^СНз СН21—1СН2
СН2
Аналогично переходят в шестичленные кольца семи- и восьмичленные и т. д. Своеобразной реакцией шестичленных нафтенов является обратимая реакция дегидрогенизации, которая приводит к образованию аро-матич. углеводородов по схеме:
н2с н2с
| сн2 | сн |
| : сн2 | Hcf>)CH |
| ^/СН2 | нс^^сн |
| сн2 | сн |
+ з Н 2
Нафтены, не имеющие шестичленного кольца, не способны к этой реакции, что м. б. положено в основу метода анализа (по Зелинскому) [8]. Однако процессы полимеризации олефинов, а также изомеризация нафтенов могут при П. п. увеличивать количество шестичленных циклов по сравнению с имевшимися в исходном продукте, а последние могут вследствие дегидрогенизации превращаться в ароматические углеводороды. Именно этим реакциям следует приписать самую сущность технологич. процесса ароматизации нефти, имеющей целью получение низших бензольных углеводородов (преимущественно бензола и а) из нефтяных продуктов. В результате дегидрогенизации могут образоваться также ненасыщенные циклич. углеводороды, обладающие подобно олефинам сильной способностью к уплотнению, к-рое может привести иногда также к ароматич. углеводородам; например Вегер при пропускании через нагретую трубку циклопентадиена получил значительные количества нафталина, образование которого можно представить по схеме:
Юн нс сн нс^ сн2
сн2
НСг"" сн
_S
НС сн
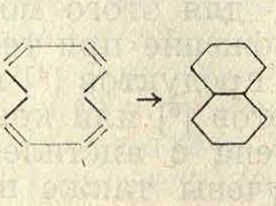
Процессы превращения нафтеновых колец при П. п. довольно хорошо согласуются с теорией напряжения Байера [9], согласно которой наиболее устойчивыми должен быть пяти- и шестичленные циклы.
4) Ароматические углеводоро-д ы являются наиболее стойкими. При не очень высоких темп-pax бензольное ядро почти не способно разрушаться, и расщепление идет преимущественно в боковых цепях, которые в отношении термич. устойчивости приближаются к цепям алифатич. углеводородов. Наиболее прочными являются короткие цепи: в частности метальная группа отщепляется от ядра при t° выше 700°. Так, Egloff и Moore [10], изучая крекинг в паровой фазе фракции ароматич. углеводородов, кипящих при 135—170°, нашли, что образование бензола совершенно не имеет места до 500° и начинается при значительно более высоких темп-pax; максимум образования а наблюдался при 750°, бензола при 800°. Наиболее характерными реакциями ароматич. углеводородов приП. п. являются превращения, связанные с уплотнением молекул. Реакции конденсации можно разбить на два следующих основных типа: 1) уплотнение ядер между собой по схеме
2СбНв->СбН6.СвН5 + Н3
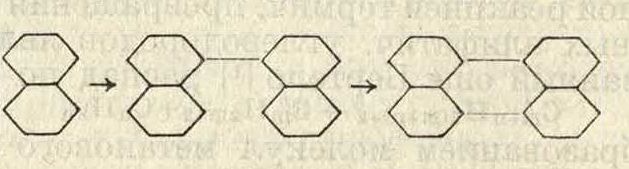
и 2) уплотнение молекул за счет ненасыщенных соединений, присутствующих в смеси с ароматическими, или за счет непредельного характера цепей у бензольного ядра. Реакция уплотнения I типа, то есть за счет подвижности водорода в бензольном ядре, весьма распространена в П. п. при высоких темп-pax; в отношении бифенила пирогенизация является даже способом его получения [п~14]. Аналогично осуществляются реакции образования динафтила из нафталина, бифенилбензола из бифенила с бензолом; динафтил ведет к образованию пирена, а также перилена [15] по схеме:
Близкими по механизму являются реакции соединения ядер при посредстве простейших боковых цепей, например при нагревании до красного каления а идет образование антрацена. Бэр и Дорп [16] получили из о-бензила также антрацен, что указывает на вероятность его роли в этом процессе как промежуточного продукта. Характерной особенностью приведенных реакций первой группы является сравнительно высокая темп-pa, необходимая для уплотнения, так как большинство представителей ароматич. углеводородов, не заключающих в боковой цепи этиленовой связи, не изменяются при t° в 450—500° [17]. Несколько иной характер имеют реакции уплотнения, идущие за счет ненасыщенных углеводородов. К таковым могут быть отнесены синтезы Berth [18], приводившие к образованию многокольчатых углеводородов при нагревании бензола и его гомологов с этиленом и ацетиленом. При нагревании этилена с бифенилом происходит образование фенан-
трена [19]. Кремер и Шпилькер [20] получили хризен из индена по следующей схеме:
сн2
CHV
VNA
% s
Данные М. Д. Тиличеева [21] по изучению сравнительной способности к коксообразова-нию углеводородов различного строения при условиях крекинга в жидкой фазе (425— 450°) показали, что наибольшую способность к реакциям уплотнения имеют соединения с двойными связями или с цепями, легко образующими таковые: инден, стиль-оен дибензил, аценафтен. Углеводороды с конденсированными ядрами без боковых цепей оказываются в этих условиях значительно более стойкими. Характерная особенность этого типа уплотнения—возможность его осуществления при более низких температурах. Поэтому есть все основания ожидать преобладающего значения их при низкотемпературных П. п., например при крекинге нефтяных продуктов, швелевании и т. д. Гидрирование ароматич. ядер в обычных П. п. имеет малое значение в виду малых концентраций Н и высокой ί° (смотрите ниже—Деструктивная гидрогенизация). В отношении химизма превращений при высокой темп-ре известно весьма немного органич. соединений, содержащих азот и кислород. Так, при температурах ниже 300° многие кислородсодержащие природные продукты (торф и др.) дают значительные количества углекислоты, которая образуется за счет отщепления С02 из карбоксильных групп гуминовых веществ. Азотсодержащие вещества каменных углей при высоких температурах коксования дают аммиак, но нек-рое количество азота остается также и в коксе.
Термодинамика П. п. Нек-рое представление о вероятности отдельных реакций, которые могут иметь место при П. п.,
можно получить из термодинамич. представлений. Согласно учению о химич. равновесии реакции протекают в сторону уменьшения свободной энергии. Изменение свободной энергии AF связано с константой равновесия Кр следующим соотношением:
In Кр - —jpf или lg Кр= - ί~ · (1)
Кр определяет собой соотношение концентраций реагирующих веществ в момент химического равновесия. Чем больше Кр, тем глубже может течь реакция и тем больше отрицательная величина свободной энергии. Величина ΔF и lg Кр позволяют т. о. судить о принципиальной возможности или невозможности осуществления данной реакции в определенных условиях. Большая положительная величина gKp указывает на реакцию, которая практически идет до конца, а при lg-KpCO реакция может пойти далеко не полно или практически не пойти вовсе. Для случаев обратимых реакций, не сопро1 вождающихся побочными процессами превращений в других направлениях, величины максимальных возможных выходов конечных продуктов м. б. точно подсчитаны по ур-иям закона действующих масс (смотрите Действующих масс закон). В приложении кП.п., где большая часть реакций не успевает дойти до состояния равновесия, применить точные расчеты не представляется возможным и потому термодинамика в данном случае дает ответ только на вопрос о принципиальной возможности тех или других реакций и отчасти дает представление о сравнительной вероятности преобладания тех или других направлений химич. превращений. Более точные расчеты позволяют сделать лишь немногие эмпирически изученные реакции, например:
С2Н6=С2Н4+Н2 [22] С,НМ=.С,Н,+ЗН2 [2 3]
СН4 =С+2Н2 [24J С2Н4 —2 С + 2 Н2 [25]
С2Н2 =2 С + Н2 [25]
Для других реакций в П. п. расчеты м. б. сделаны лишь грубо приближенно. Неточ-
Т а б л. 1.—Р еакции расщепления параф и, новых углеводородов.
| Реакции | Тепловой эффект Q в cal | lgKp при | 1° | |||
| 400й | 500° | 600° | 700° | 800° | ||
| СНД ^ С 4- 2Ы2.
С4Н10 С4Н9 + Н2. С4Н10 сы4 + С3Н«. С4Н10 <± с2нв + С2Н4. СвН]4 ^ΟβΗΐ2 + Η2. С6н14 ^ СН4 + СбНю. CeHi4 СзНз 4- СзНг,. С6Н14 ^ С4Ню + С2Н4. CioH22;±c6Hi2 + С5Н10. Ci2H26 ^ СНд + СцН22. ОцНйв^СвН,4 + C6Hi2. C12H2g^CioH22 + C2H4. С/20-И-42 Ci(,H22 + C10H20 · · · |
-18,000 -32,000 -25,000 -26,000 -32,000 -24,000 -32,000 - 41,000 -29,000 -24,000 -22,000 -32,000 -12,000 | -0,1 -3,9 -0,6 -0,5 -3,9 -0,4 -2,5 -5,4 -1,5 + 0,4 +0,8 -2,5 + 4,0 | +0,8 -2,6 + 0,5 +0,7 -2,6 +0,8 -1,2 -3,6 -0,2 + 0,8 +1,8 -1,2 +4,7 | + 1,4 -1,3 + 1,4
+ 1,6 -1,3 + 1,6 +0,1 -2,1 +0,9 +1,6 + 2,6 + 0,1 + 5,1 |
+2,0 -0,4 + 2,1 +2,4 -0,4 + 2,3 + 1,0 -1,0 + 1,7 +2,3 + 3,3 + 1,0 + 5,5 | + 2,3 +0,4 +2,7 + 3,0 + 0,4 + 2,9 + 1,8 0,0 + 2,4 + 2,9 +3,8 + 1,8 + 5,9 |
| T а б л. 2 | — Реакции расщепления олефипов. | |||||
| Реакции | Тепловой эффект Q в cal | IgKp при 1 | ||||
| 400° | 500° | 600° | 700° | 00
о о | ||
| с»н, А с -г сн4. | +35,000 | 16,0 | 14,0 | 13,0 | 12,0 | 12,0 |
| С4Н» Ϊί 2С2Н4. | -41,000 | -5,4 | -3,6 | — 2,1 | -1,0 | 0,0 |
| Cigli20 2С5Н10. | -30,000 | -1,8 | -0,5 | + 0,6 | + 1.5 | + 2,2 |
Таблица 3.—P e а к ц и и расщепления ароматических углеводородов.
| Реакции | Тепловой эффект Q в cal | lg Кр при t | 0 | |||
| 0
0 |
500° | 600” | 700° | 800° | ||
| СвН5-СгН5^:СвНв + C2Hi. | -35,000 | -3,5 | — 1,9 | -0,7 | +0,3 | +1,1 |
| ОвНб“СзН7 СбН6 +- С3Нв · · · | -27,000 | -0,8 | +0,4 | 4-1,5 | +2,2 | +2,8 |
| С6Н5-С3Н75>СвН6-СН3 + С2Н4 | -31,000 | -2,0 | -0,7 | + 0,4 | +1,2 | +2,0 |
| СвНб“С4Н9 ΟβΗβ + С4Н8. ·. | -29,000 | —1,4 | -0,1 | +0,9 | +1,7 | + 2,4 |
| СвН5-С4Н9 д7СвН5СН3 + С3Н6 | -28,000 | -1,1 | +0,2 | -1,1 | + 1,9 | + 2,6 |
Таблица 4.—Р аспад бензольного ядра.
| Реакции | Тепловой эффект Q в cal | lg Кр при t | ||||
| 40(Т° | СЛ
О о о |
600° | 700° | 800”
i | ||
| С6Нб СН4 + Н2 + 6 С. | +29,000 | 15,5 | 14,4 | 13,5
14,8 |
12.9 | 12,3 |
| С0Нв Д7 6С + 31Г2. | + 11,000 | 15,3 | 15,0 | 14,7 | 14,6 | |
Таблица 5.—Р еакцпи кольцеобразования.
| Тепловой | lg Кр при | |||||
| Реакциа | эффект | ---;- | ||||
| Q в cal | 400° | 500° | 600° | О
О с- |
СО
о о | |
| Гексилеп ;± циклогексан. | + 15,000 | +4,9 | +4,2 | +3,8 | +3,4 | +3,0 |
| Амилен τι циклопентан. | + 19,000 | +6,2 | + 5,4 | + 4,8 | + 4,3 | + 3,9 |
Т а б л. 6.—Д егидрогенизация нафтенов.
| Реакции | Тепловой эффект Q в cal | lg Кр при 1“ | ||||
| 400° | 500° | 600° | о о
о |
о о со | ||
| C6Hi2 С6Н6 + ЗН2.
С6НдСНз^СвНб*СНз + зн2 CioHis*1 C1oHe + 5Н2. CioHi2*2 CioHg + 2Н2. |
-54,000
-49,000 -74.000 -30,000 |
+ 2,2 +3,7 +8,7 +3,4 | + 4,7 + 6,1 +12,3 + 4,8 | + 6,7 + 8.0 + 15,2 + 6,0 | + 8,4 + 9,5 + 17,5 + 6,9 | + 9,7 + 10,7 + 19,5 + 7,7 |
*ι Декалии. *2 Тетралин.
Таблица 7.—У п л о т н е н и е ароматически ность расчетов усугубляется недостаточностью и недостоверностью имеющихся в литературе величин тепловых эффектов реакций и других необходимых данных. В табл. 1—7 приведены величины lg Кр для нек-рых реакций, имеющих место при П. п., которые м. б. вычислены на основании имеющихся в литературе данных для тепловых эффектов реакции по приближенному ур-ию Нёрнста:
lg Кр=- Ш1,75 lg Т - τηΟ, (2)
где Q—>тепловой эффект реакции, ~Ση—алге-браич. сумма молекул, участвующих в реакции, С—химич. постоянная. Величины химич. постоянных приняты приближенно Сц2 =1,6, Ссп4=2,5 и для других углеводородов Суы.=3,0 [26].
Из этих расчетов по величине и знаку lg Кр
х и непредельных углеводородов. можно судить о том, при каких температурных условиях какие реакции термодинамически возможны и насколько полно они могут протекать.
Помимо ур-ий Нёрнста для суждения о возможности течения при П. п. различных реакций (для приближенных расчетов) м. б. применено ур-ие (3), выражающее зависимость AF от темп-ры при реакциях образования углеводородов из элементов:
AF=АН — Т AS, (3)
где АН—тепловой эффект реакции, a AS— изменение энтропии при реакции [26]; см. табл. 8.
Из приведенных в табл, ур-ий по свойству аддитивности AF можно рассчитать изменение свободной энергии при различных пи-рогенетических реакциях. Применяя ур-ие (1) возможно рассчитать величину lg Кр, а следовательно и охарактеризовать термоди-
| Реакции | Тепловой эффект Q в cal | lg Кр при t | ° | 1 | ||
| 0
0 |
500° | аз о о | о о
t> |
800° | ||
| 2С6Нв^:СвН6-СбН5 + Н2. | + 1,000 | -1,1 | -1,1 | -1,2
+5,4 | — 1,2
+5,8 |
-1,2 |
| 2СбНв + С2Н4 ^ С14Н10*1 + ЗН2С6Н5.С5Ы5 + С2Н4 £ Οι4Η10*2 + | + 6,000 | +5,7 | +5,5 | +5,3 | ||
| + 2Н2.. | +9,000 | + 8,1 | +7,8 | + 7,6 | +7,5 | + 7,3 |
Антрацен. *2 Фенантрен.
Таблица 8.—3 начения величины &F при реакциях образования углеводородов из злементов.
| Реакции | Δ F |
| С + 2 Н2 — CII4 | - 20 000 + 26,2Т |
| 2 С + ЗН2=СгНв | - 29 000 + 53,ЗТ |
| 8 С + 9Н2=CgHig | - 76 100 + 206,6/· |
| п С + (п+1) Н2=СцН2п+2 | - 13 300 - 7 850п + 2,2Т + 25,55пТ |
| а с + 2на - с2н4 | + 12 600 + 11,6Т |
| п С + п Н2=СпН2п | + 31 060 - 9 400п - 39,5Т + 25,55пТ |
| 2 с + н2 - С2Н2 | + 54 400 - 10,8Т |
| пС + (п — 1) Н2=СпН2П-2 | + 68 400 - 7 ОООп - 61.9Г + 25,55пТ |
| 6 с + зн2=свнв | + 17 000 + 35,ЗТ |
| 7 С + 4 Н2=СбНз-СНз | + 6 860 + 59,2Г |
| 6 С + 6Н2=C6Hi2 (циклогексан).. | - 38 100 + 134,ОТ |
| 10 С+ 4Н2=С10Н8 (нафталин) .. | + 28 100 + 47,5 Г |
| nC+ пН2 - СпН2п (нафтен) | + 12 900 - 8 500η - 19,3Г + 25,55пТ |
намич. возможность осуществления данной реакции. В заключение следует отметить, что полной картины П. п. термодинамика дать не может, т. к. при всех 1° пирогенетич. разложения углеводородов наиболее полно может идти глубокий распад до СН4, Н2 и С, что конечно и может иметь место на практике при достаточно длительном ведении процесса. Однако наибольший практич. интерес имеют процессы, ведущие к образованию промежуточных продуктов, и потому для описания П. п. кроме термодинамич. представлений необходимо привлечь также кинетические. Только знание законов изменения скоростей отдельных реакций позволит ска-
молекулы углеводорода. Экспериментальные и теоретич. исследования в этой области не дают еще достаточного материала для полного уяснения процесса термич. разложения; выяснено, что увеличение длины цепи углеводородного соединения приводит к увеличению константы Си следовательно к соответственно большей скорости распада; цик-лич. же структура, наоборот, увеличивает сопротивляемость распаду и тому подобное. Что же касается величины энергии активации, то (поскольку можно судить по расчетам на основании не вполне точных экспериментальных данных последних лет) она s 65 000 cal и не зависит от величины молекулы (смотрите табл. 9).
Таблица 9.—Э нергия активации термического распада углеводородов.
| Вещество | Условия процесса разложения | Е 10-3 в Cal | Авторы |
| 1 Пропан и бутаны.
1 Грозиелский параф, мазут. Грозпенский параф, дистнл- лат.. Парафин .. :Рангоунский парафин. |
Разложение в паровой фазе при 600—650° Крекинг в жидкой фазе при 400—450°
» ·> » » » 375—475° » » » » » 425—450° » » » » » 450—460° |
65 + 3 63 ± 14
64+8 64.5 67.5 |
Pease,
Durgan I28] Саханов и Тиличеев [26) » » Waterman, Perquin [9»j |
зать, какие из термодинамически возможных реакций получат наибольшее развитие.
Кинетика П. и. и катализ. Большинство реакций, имеющих место в П. п., являются необратимыми, так как первичные продукты распада немедленно претерпевают дальнейшее превращение и неспособны уже дать снова исходный продукт. В виду этого динамика превращений в П. п. в значительной мере определяется соотношением скоростей отдельных реакций. Изучение кинетики П. п., начавшееся лишь в самые последние годы, в значительной мере может помочь в деле выяснения роли различных факторов и обусловить возможность полного управления процессами. Что касается первичной реакции П. п. термического разложен и я, то, как это следует из новейших исследований [2e,27J, оном. б. признано процессом гомогенным и мономолекулярным, скорости которого не зависят от давления и являются ф-ией темп-ры:
Б
И^,= Ce~RT,. (4)
где Е—энергия активации реакции расщепления (смотрите Кинетика химическая) и С—константа, зависящая от величины и строения
Большая величина энергии активации распада углеводородов также приводит к заключению, что реакции термич. разложения не м. б. причислены к типичным каталитич. реакциям (смотрите Катализ). Значительно менее ясен характер процессов уплотне-и и я, всегда сопровождающих П. п. Однако в первом приближении и они м. б. в условиях обычных П. п. признаны преимущественно гомогенными. Будучи бимолекулярными, реакции полимеризации и конденсации в значительной мере зависят от давления; их скорости подчиняются ур-ию:
_:ji-
Wyn.t=kABe R T, (5)
где.А и В—концентрации уплотняющихся молекул. Ли В являются ф-иями давления, и ур-ие (5) м. б. представлено в виде:
Е
Wynn.=kP}РБе~ RT, (6)
где РА и Рв—парциальные давления реагирующих веществ. Экспериментальные данные [30J указывают на увеличение скорости полимеризации этилена с повышением давления. То же следует из данных о влиянии давления на непредельность бензинов при крекинге [26]. Величина энергии активации процессов уплотнения также не вполне выяснена. Экспериментальные данные Реаве[зл], полученные при изучении кинетики полимеризации олефинов, указывают на величину J3=35 000 cal. Переходя к реакциям дегидрогенизации, имеющим значение для технич. процесса ароматизации, следует признать таковые в значительной степени гетерогенными. Что касается обратной реакции гидрирования, то в обычных П. п., где парциальное давление водорода в газах весьма мало, она практического значения не имеет. Процессы дегидрирования шестичленных полиметиленовых колец с образованием ароматич. углеводородов показали, что присутствие катализаторов в значительной мере способствует этому процессу. Катализаторами для дегидрогенизации являются все гидрирующие металлы, к каковым относятся Pd, Pt в качестве наиболее нежных, работающих при 1° ниже 300°, и Ni, Со, Fe и др., осуществляющие отщепление водорода при более высоких темп-рах. Хотя в технике ароматизацию ведут при темп-рах выше 600° и без применения специальных катализаторов, но так как эта реакция в сущности является реакцией дегидрирования шестичленных нафтенов, то ее можно считать каталитической.Роль катализатора в этом случае играют железные стенки реторты, в которой производится процесс. В виду этого использование в технике чисто каталитич. дегидрогенизации соответствующих фракций нефтей для получения наиболее ценных технич. продуктов (бензола и а), взамен дорогой и связанной с· большими потерями ароматизации нефтяных дистиллатов, принципиально вполне возможно; единственным препятствием пока можно считать отсутствие вполне подходящего и дешевого катализатора.
Деструктивная гидрогенизация. Если обычные П. п. складываются из двух основных реакций: термин, распада и уплотнения молекул, то деструктивная гидрогенизация, или крекинг, в присутствии водорода характеризуется одновременным сн сн нс+Х^Чсн „ :i |с сн
НС /с С СН -
сн С| ! г1
H<V+>H
сн нс
II I
/Л. II
+ .1 II I
ч и т. д.
Процесс сопровождается образованием больших количеств метана и этана. Исходя из этого следует, что в качестве исходного материала наиболее выгодными для получения максимального выхода легких бензиновых углеводородов должны быть продукты преимущественно парафинового основания,то есть не содержащие больших количеств сложных ароматич. полициклических углеводородов, т. к. последние для превращения в легкие углеводороды требуют большой затраты водорода и дают весьма большие количества малоценных углеводородных газов (метан, этан) при малых выходах легких жидких углеводородов, то есть бензина. Наибольший интерес для овладения процессом деструктивной гидрогенизации представляет управление реакцией присоединения водорода. Эта реакция при достаточном своем развитии может полностью устранить процессы уплотнения, следовательно и коксообразова-ние. Как показали последние исследования |зз—86j и описания заграничных установок, для достижения достаточного эффекта необходимо значительное повышение давления водорода. Анализ роли давления [37] при процессе сводится к двум основным моментам.
1. Равновесие гидрогенизации. Рассмотрим влияние повышения давления в отношении двух основных типов соединений: непредельных и ароматических углеводородов. И те и другие в наибольшей степени способны к реакциям уплотнения и могут обусловливать коксо-образование, что и является главным затруднением в осуществлении процесса. Для характеристики состояния равновесия реакций гидрирования олефинов при различных условиях более точные данные имеются только для этилена [**]. В табл. 10 даны
Таблица 10.—Р авновеспе гидрирования этилена (С2Н4+Н2 С2Н0).
| г | • lg Кр | ί рспн2п карсан2„+2 Рп2 | |||||
| Эксперимен тально | Вычислено но Френсису | рн2=1 | atm | Рн2 = 50 atm | |||
| п=2 | п=2 | п =8 | П—2 | n—S | η =2 | η—8 | |
| 400
450 500 |
-3,84
-3,14 -2,53 |
-4,3
-3,3 -2,5 |
—1,2 -0,5 +0,1 | 0,00005
0,0005 0,0032 |
0,06
0,3 1,3 |
ι,ο·ιο-β
1,0-10-5 6,4-10-5 |
0,0012 0,0064 ! 0,025 |
наличием третьей реакции—гидрирования. Химизм процесса заключается в том, что к продуктам распада ненасыщенного характера присоединяется водород и препятствует наступлению полимеризации. Кроме того в результате гидрирования ароматич. колец сложных полициклич. соединений образуются более легкие углеводороды. Н. А. Орлов Г12] для химизма превращений, происходящих в условиях деструктивной гидрогенизации хризена, дает следующую схему:
результаты подсчета по ур-ию (7), выведенному из расчетов Френсиса [25] (смотрите табл. 8):
lg Кр=— 9 + ^г-п+ 9,13. (7)
Для характеристики степени точности этого грубо приближенного расчета в таблице 10 приведены данные, вычисленные на основании эмпирического уравнения (8) Pease и Durgan [22J для гидрирования этилена
lgK?=-^+6,31. (8)
В отношении гидрирования ароматич. углеводородов экспериментальные данные также весьма бедны, и более точный расчет возможно сделать только для системы бензол— циклогексан: CeH„ + 3H2?CeHt2. Результаты подсчета на основании эмпирически полученного уравнения Берроуз-Лукарни [23]
lgiri,=-12T050 + 21,59 (9)
приведены в таблице 11, в которой рсвн„
Кр =
рк
Л =
рс„н«
рс„н,
(10)
(из ур-ия 10) изменяется в зави-водорода.
РС6Н _
симости от темп-ры и давления нием водорода к продуктам реакции. Если скорость гидрирования относительно мала, то процесс имеет характер обычного крекинга, если же, наоборот, она велика, то продукты уплотнения не образуются вовсе. Таким образом получение тех или других продуктов зависит от возможности регулирования скоростей реакций и в частности реакции гидрирования. Скорость гидрирования (ТТшйр.) должна удовлетворять ур-ию Аррениуса:
_ Е
WmdP=KCy,.i. -Сщ.е т (И)
где Сугл. и Сд2—концентрации углеводородов и водорода, Е—энергия активации реакции и К—константа, зависящая от приро-
Т а б л. И.—Р авновесие гидрирования бензола.
| 1 рсвнв К | Р | ||||||||
| t° | К п | рсвн1а р | 3
Н 2 |
||||||
| Р | рн2= | 1 atm | РН2= =50 atm | Pn2=ioo airn | рн | 2=200 aim | Рд2=500 atm | ||
| 400 | +3,69 | 4,9-10* | 4,9 | 103 | 0,039 | 0,0049 | 0,0006 | 3,9-10-6 | |
| 425 | +4,33 | 2,1·104 | 2,1 | 104 | 0,17 | 0,021 | 0,0026 | 0,00017 | |
| 450 | +4,92 | 8,3104 | 8,3 | 104 | 0,66 | 0,083 | 0,0104 | 0,00066 | |
| 475 | +5,48 | 3,0-10« | 3,0 | 10« | 2,4 | 0,30 | 0,038 | 0,Q024 | |
| 500 | +6,00 | 1,0-10«
1 |
1,0 | 10е | 8,0 | 1,0 | 0,125 | 0,008 |
Для более сложных ароматич. углеводородов пока данных нет, а наиболее важные для понимания процессов глубокого уплотнения высококонденсированные ароматические углеводороды настолько мало изучены, что не представляется возможным сделать даже приближенные расчеты. Однако порядок величин вероятно близок и потому нет оснований предполагать здесь принципиальное отличие. Несмотря на приближенный характер сделанных выше расчетов, можно с несомненностью сделать вывод относительно большей полноты, при тех же условиях, гидрирования олефинов, чем это имеет место в отношении ароматич. углеводородов. Для гидрирования высокомолекулярных углеводородов не следует значительно повышать темп-ру, т. к. состояние равновесия при этом сдвигается в сторону обратной реакции. Последнее заключение находит себе подтверждение как в экспериментальных результатах лабораторных исследований, так и в описаниях заграничных установок, которые для гидрогенизации тяжелых продуктов не повышают 1° более 425—450°, а во второй фазе, где преобладают легкие фракции, поддерживают ί° до 490—500°. Однако, как это следует из экспериментальных данных лабораторных исследований [83~37], процессы гидрогенизации не приводят к состоянию равновесия и в продуктах гидрогенизации присутствуют не только ароматические, но даже низшие непредельные углеводороды, которые должны были бы полностью гидрироваться уже при низких давлениях водорода. Это объясняется скоростью гидрирования.
2. Скорость реакций и давление водорода. Если при обычном крекинге процесс протекает гл. обр. в двух направлениях: расщепления и уплотнения, то деструктивная гидрогенизация характеризуется реакцией третьего типа—присоедине ны углеводородов. Для случая поАюянной
Е
1° член e RT м. б. введен в константу, и тогда получим следующее ур-ие:
^гидр. = & CW.C1j2 (12)
или, заменяя концентрации парциальными давлениями Ругл_ и Ря, имеем
Wmdp.=КРуглРщ, (1.1)
откуда следует, что увеличение давления водорода также в значительной степени вли-яет^на скорость присоединения водорода. Количественно это положение подтверждается экспериментальными данными лабораторных исследований [*7]. Однако уравнение (13) оказывается справедливым только для гомогенного проведения реакции в паровой фазе. В случае жидкофазной гидрогенизации, при малой растворимости водорода в углеводородах (особенно при высоких температурах), необходимо создать специальные условия для увеличения поверхности соприкосновения продукта с водородом. К таковым относится прежде всего энергичное перемешивание, без которого процесс идет весьма несовершенно. Кроме того в жидкофазном процессе катализатор, помимо своей прямой роли, а именно уменьшения энергии активации реакции, служит также передатчиком водорода в жидкую фазу: частицы катализатора, попадая при перемешивании из паровой фазы в жидкую и обратно, переносят адсорбированный на своей поверхности водород в жидкость.
Лит.: i) Berthelot М., «Annales de chimie et de physique», P.,1866,t-9,p.455; «Mechanique chimique», P., 1879, t. 2, p. 119; 2) HurdC., Spense L., «Journal of the American Chemical Society», Easton, Pa., 1929, v. 51, 11, p. 3353; з) Ипатьев В. Н., Полимеризация олефинов, «Ж», 1906, т. 38, стр. 63; 1911, т. 48, стр. 1420; *) Бызов Б., Сов. П. 6321, Способ получения диолефиновых углеводородов; 6) Лебедев С., Сов. П. 24393, Получение искусствен-
ного каучука; e) Plauson’s Forschungsinstitut, Г. П. 338030, Способ получения дивинила; 7) Розанов И. А., Исследования в области изомерных превращений циклич. соединений, стр. 12, М., 1916; «) Работы академика Н. Д. Зелинского и его сотрудников, «Ж», 1911, т. 43, стр. 1220; 1912, т. 44, стр. 275; «В», 1912, т. 45, стр. 3678; 1923, т. 56, стр. 716 и 1723; 9) Меншуткин Б. Н., Карбоциклич. соединения, стр. 13, Л., 1926; м) E g 1 о f f a. Moore, «Industrial a. Engineering Chemistry», Wsh., 1917, v. 9, p. 40; n) Bertbelot V.,«Ztschr. f. Chemie», 1866, p. 707; 1Z) Meyer H., Hofmann A., «Monatshefte f. Chemie», W., 1916, B. 37, p. 681; >«) Z a n e 11 i J., Egloff «Industrial a. Engineering Chemistry», Wsh., 1917, v. 9, p. 350; ы) LoweC., James C., «Journal of the American Chemical Society», Easton, Pa., 1923, v. 45, p. 2666; is) Scholl, Weitzen-beck, «В», 1910, B. 43, p. 2203 ; i«) Behr u. Dorp, «В», 1873, B. 6, p. 754; 17) Добряпский А. Ф., Пирогенетич. разложение нефти, стр. 24, Л., 1922; 18) В erth, «CR», 1866, t. 63, p. 792, 835, 999; i») В а г b i er, «Annales de Chimie et de Physique», P., 1876, siSrie 5, t. 7, p. 532; м) К r a m e r, S p i 1-k e r, «B», 1890, B. 23, p. 3276; m) Ти л иче ев М. ДБ, Доклад на конференции по крекингу и гидрогенизации в г. Грозном, 1931; 22) Pease and D u r-g a n, «Journal of the American Chemical Society», Easton, Pa., 1928, v. 50, p. 2715; Berrous, -Lucain i, ibid., 1927, v. 49, p. 1157; 24) Egloff,
S r. h a a d, Lowry, Литературная с «The Journo I of Physical Chemistry», Ithaca, N. Y., 1930, y. 34, p 1617; 25) f r a n c i s, «Industrial a. Engineering Chemistry», Wsh., 1928, v. 20, p. 277; 2в) с ахая о в
A. H. и Тил иче e в М. Д., Крекинг в жидкой фазе, стр. 153, М.—Л., 1928; 27) L e s 1 i e, P о t h о i f. «Industrial a. Engineering Chemistry», Wsh., 1926, S; 2») P e as e, D u rga n, «Journal of the American Chemistry Society», Easton, Pa., 1930, v. 52, p. 1262; 28) Waterman, P e r q u i n, «Journal of the Institution of Petroleum Technologists», London, 1925, v 11, p. 45; 30) и π a t ь e в В. H. и Немцов M. С.,
О получении бензинов и газов крекинга, 1931; 31) Pease В. N., Kinetics of the Polimerization of Ethylene at Pressures above one Atmosphere, «Journal of the American Chemical Society», Easton, Pa., 1931, v. 53, p. 613; 32) Орлов H. А., Сжижение угля и его продуктов, стр. 19, М., 1930; si) Ипатьев
B. Н., Б е л о π о л ь с к и и М. А. и Немцов М. С., Крекинг грозненского парафинистого мазута в присутствии водорода, «Труды конференции но крекингу и гидрогенизации в г. Грозном», 1931; ««) С а х а-п о в А. Н. и Тиличеев М. Д., Гидрогенизация нефтяных продуктов, там же, 1931; зз) Waterman, p e r q u 1 n, «Journal of the Institution of Petroleum Technologists», London, 1925, v. 11, p. 36; 3«) К ling A., Florent in D., «Bulletin de la Societe chimique de France», P., 1927, t. 10, p. 864; 37) H e μ ц о в M. С. и Ф p о c t А. В., Гидрогенизация нефтяных продуктов, «Труды конференции по крекингу и гидрогенизации в г. Грозном», 1931; Naf-t а 1 i M., Standard J. G. Со., «Ztschr. i. angewandte Chemie», B., 1930, Jg. 43, p. 58. IVI. Немцов и А. Фрост.