 > Техника, страница 74 > Промежуточные продукты
> Техника, страница 74 > Промежуточные продукты
Промежуточные продукты
Промежуточные продукты для синтеза красителей являются переходной ступенью от простейших углеводородов (смотрите Коксобензольное производство) к сложным красителям (смотрите Красящие вещества синтетические) и производятся на анилокрасочных ф-ках в значительных количествах и в большом многообразии. Нередко получение красящих веществ из П. п. не представляет технич. затруднений, в то время как изготовление П. п. требует ряда сложных операций, протекающих в сложных технич. аппаратах. Так, азокрасители (смотрите) получают при помощи сравнительно простой операции азосочетания диазотиро-ванных аминов с производными аминов и фенолов бензольного или нафталинового ряда; эти же П. п. для синтеза азокрасителей получают путем последовательного ряда сложных операций, которые будут описаны ниже. Точно так же весьма ценные и сложные по составу кубовые красители (смотрите) получаются при несложной конденсации заранее приготовленных П. п. индольного и антрахиноно-
вого ряда, изготовление которых представляет значительные технич. затруднения. Значение П. п. в технологии красящих веществ весьма велико. Кроме того П. п. нередко являются конечными продуктами или П. п. для фармацевтич. и фотографич. препаратов, чатых и отравляющих веществ. Основным органич. сырьем для производства П. п. служат циклич. углеводороды ароматич. ряда и нек-рые гетероциклич. соединения, например бензол, ксилолы, нафталин, антрацен, карбазол и др., получаемые из каменноугольной, коксовой или газовой смолы, или при пирогенизации нефти. Эти циклич. соединения, подвергаясь ряду последовательных операций, переходят в П. п. различной степени сложности. П. п. не могут быть классифицированы по своим функциям, т. к. весьма часто один и тот же П. п. обладает различными группами, обусловливающими различные функции. Для систематич. ознакомления с П. п. необходимо в силу этого рассматривать в определенной последовательности не самые П. п., а те основные химии. реакции, при помощи которых эти П. п. получаются.
Классификация этих реакций м. б. произведена по следующим признакам. В первую очередь все реакции м. б. разбиты на две группы: первая из них, большая по объёму, объединяет все реакции конденсации (смотрите), в которых два остатка, (радикала) двух химии, соединений вступают в тесную, «плотную», связь, образуя продукт конденсации. В случае, если оба вступающие в конденсацию остатка находятся в одном и том же соединении, имеет место внутримолекулярная конденсация, приводящая либо к новому, по сравнению с исходным, взаимному положению групп, называемая п e р е г р у п-пировкой, либо к замыканию нового цикла, называемая циклизацией. Вторая группа реакций представляет реакции восстановления и о к и с л е н и я; в этой группе также возможны внутримолекулярные реакции, если окисляющая и окисляемая группы находятся в одном и том же соединении. Реакции конденсации в свою очередь м. б. разбиты на два класса. В первом из них конденсация обусловлена прежде всего ароматич. характером соединения, вступающего в конденсацию, и приводит к введению в ядро циклич. соединения новой группы. Во втором классе—конденсация обусловлена гл. обр. специфич. свойствами групп, представляющих боковую цепь вступающего в конденсацию соединения, а не его ароматич. свойствами. Все же в силу наличия этих ароматич. свойств продукты конденсации второго типа в боковой цепи соединения способны к внутримолекулярной перегруппировке, когда введенная в боковую цепь группа перемещается в ядро соединения. К первому классу конденсаций относятся реакции введения (в ароматич. ядро) нитрогруппы, сульфогруппы, галоида и углеродсодержащих групп, а также введение свободных и замещенных гидроксильной и амидной групп путем обмена заранее имевшихся нитро-, сульфо-, амино-, окси-групп и галоида. Ко второму классу конденсаций относятся реакции гидроксильной, амидной, альдегидной, метильной и нитрозо-групп, обусловленные в первую очередь реакцие-
способностью этих групп, а не ядра соединения. Т. о. все реакции получения П. п. могут быть разобраны по нижеследующей классификации.
I. Реакции конденсации.
A. Реакции, обусловленные ароматич. характером соединения:
1) нитрование,.,
2) сульфирование,
3) галоидирование,
4) аминирование и гидроксилирование,
5) введение углеродсодержащих групп.
Б. Реакции, обусловленные свойствами боковой цепи соединения:
1) ацилирование,
2) алкилирование,
3) введение в боковую цепь двувалентных метиленовой и амидной групп.
B. Реакции внутримолекулярной конденсации:
1) циклизация,
2) перегруппировка.
II. Реакции окисления и восстановления.
Весьма часто мы имеем дело со сложной реакцией, которая для правильного понимания должен быть расчленена на ряд последовательных реакций, относящихся к различным классам. В дальнейшем реакции получения П. и. рассматриваются в последовательности, соответствующей вышеприведенной классификации, с попутным разбором наиболее характерных и ценных из получаемых по этим реакциям П. и.
I. Реакции конденсации. Они м. б. представлены в наиболее общем виде схемой R ·×+ В,! · Y -► R · Ri +×· Y,
к-рая показывает, что остатки R и Rx в результате конденсации пришли в плотную связь. Реакции конденсации, обусловленные ароматич. характером одного из вступающих в конденсацию соединений, должен быть рассмотрены более детально. По К. Мейеру ароматич. свойства соединения выражаются в активности двойной связи этого соединения, и можно представить, что в первую очередь по месту этой двойной связи идет присоединение элементов второго соединения, вступающего в конденсацию. Затем при наличии надлежащих условий (водо- или кислотоотнимающие средства, повышенная Г, катализаторы и тому подобное.) из продукта присоединения удаляются элементы воды, кислоты и т. и. с образованием ароматич. соединения, содержащего новую группу. Сказанное можно выразить схемой
ож
/Ri
R
/X
Н
/Ri
+ RX
По этой схеме реакции нитрования (I), сульфирования (II), галоидирования (III), ами-нирования (IV), гидроксилирования (V) и ввода углеродсодержащей группы (VI) м. б. изображены следующим образом
#-ч н н
/Н
no2
он
3*4
no2
/ОН
Ч/Ч-
/ч
no2
Н20
^4j/ S03H
+ I -
^ч/:
ч он
I.
Б03н н
/ОН <
ч/ч-
✓ч ч
so3H
+ н20
^ЧУ
ч/ч
/Н
н
f
1 н
/4/R
% и
/ч/н
Ч н
N.
ч
С1
С1
а,
R2
4#
I 1
I Г/С1-% нIII.
,
м
. · ЧП
; /х -^/Чн IV
/Ч
Ч
CI
НС1
/4_N/R>
4r2
ч/1
RX
О— R,
I
X
/II
cfR
R
/4/°-R> /4/°“^
/л
Η
V.
/R
/CfR
< XK
XH
/x 4 H
VI.
4
4x C^R
• I I Xr-
I il
4
IIX
Существование продуктов присоединения с бесспорностью доказано для реакций галоидирования, для остальных же реакций их существование не противоречит, а иногда и подтверждается всем ходом реакции. Удаление из продуктов присоединения элементов воды, НС1, RX и тому подобное. происходи! под влиянием водоотнимающих средств (для удаления воды), повышения f° или катализатора (для удаления НС1 и избытка вступающего в реакции агента—в случае гидроксилирования и аминирования). Гидролиз сульфогруп-пы в сульфокислотах (смотрите ниже) также является нек-рым подтверждением промежуточной стадии присоединения, но реакция в случае гидролиза, то есть при недостаточной крепости водоотнимающего средства, идет в обратном направлении. За последнее время предложено объяснение ароматич. свойств соединения электронными явлениями, но до сих пор нет еще вполне четкого представления в этой области, и мы будем в силу этого придерживаться вышеприведенных взглядов об активности двойной связи и о промежуточной фазе присоединения. Степень легкости конденсации,обусловленной ароматич. свойствами соединения, или, иначе говоря, степень легкости введения в ядро ароматич. соединения вместо водородного атома или какой-либо новой группы, зависит от активности двойной связи и от подвижности водородного атома или уже имеющейся группы. Активность двойной связи облегчает присоединение по ней и образование промежуточной стадии—продукта присоединения
/4/Rl
4R
:!/х
ч н
Подвижность же водорода или группы R облегчает выделение из этого продукта молекулы RX. В незамещенном бензоле мы имеем равноценные двойные связи и равноценные атомы водорода. В производных же бензола заместители могут оказывать двоякое влияние. Одна категория заместителей облегчает реакцию конденсации, направляя в то же время новую группу в о- или п-положение < к уже имеющемуся заместителю; вторая ва-
тегория заместителей затрудняет конденсацию и, препятствуя введению заместителя вообще, препятствует в меньшей мере введению его в ж-положение к уже имеющемуся заместителю. К первой категории относятся группы ОН, NHj, галоиды, СН3 и их производные, ко второй—S03H, N02, а также СООН и другие карбонилсодержащие группы с карбонилом при ядре. В двузамегценных бензолах возможны случаи, когда оба заместителя направляют согласованно, облегчая тем самым конденсацию, или когда оба заместителя направляют в разные места, образуя при этом вследствие конкуренции смесь изомеров, причем превалирует в этой смеси тот изомер, который получился вследствие направляющей роли более активного заместителя. В нафталиновом ряду в отличие от бензольного даже в незамещенном нафталине не все водородные атомы равноценны. Так, водородные атомы в α-положениях более подвижны, чем в ^-положениях, вследствие чего конденсации в нафталиновом ряду идут в первую очередь в α-положении. В замещенных нафталинах наблюдаются те же закономерности, что и в бензольном ряду, но число возможных «хиногенных» мест, то есть равноценных о- и и-местам бензола, значительно больше. Так например, для а-заме-щенных нафталина
тm
а а 5 4
хиногенными по отношению к месту 1 будут положения 2, 4, 5, 7, причем легкость вступления новой группы в хиногенные места убывает в последовательности положений 4, 2, 5, 7. Для ^-замещенных нафталина хино-генными по отношению к месту 2 являются положения 1, 3, 6, 8, причем легкость вступления новой группы в хиногенные места убывает в порядке положений 1, 8, 6, 3. (В главе о сульфировании нафтолов будет приведена полная иллюстрация этого явления.) В антрацене наиболее реакциеспособными
Н У 1
зОООс
5 10 4
Антрацен я СО 1
7 6
5 СО 4 Антрахинон

2
3
являются мезо-положения 9 и 10, куда преимущественно и направляется присоединение или конденсация. Но иногда, в частности при сульфировании, антрацен ведет себя как замещенный нафталин, реагируя в первую очередь своими α-местами (1, 4, 5, 8), а во вторую очередь /3-местами (2, 3, 6, 7). В ан-трахиноне мы имеем два равноценных бензольных ядра, связанных двумя карбонильными группами. Поэтому получение монопроизводных антрахинона представляет некоторые трудности, т. к. каждое из бензольных его ядер способно к реакциям присоединения. Указанные общие положения о конденсации, обусловленной ароматич. характером соединения, позволяют перейти к рассмотрению отдельных типов этих реакций.
А. Реакции, обусловленныеаро-матическим характером соединений. 1) Реакция нитрования согласно вышеизложенному м. б. выражена следующей схемой (для бензола)
no2
он
А
NO,
/NO 2
Н
н он
+ н20
Как видно из приведенной схемы, для доведения реакции до конца необходимо наличие водоотнимающего средства. Действительно в отличие от жирных углеводородов, которые нитруются разбавленной азотной кислотой при повышенных температуре и давлении, ароматические углеводороды и их производные нитруются крепкой азотной кислотой при участии водоотнимающих средств, преимущественно серной кислоты, и при более низкой температуре (смотрите Нитрование). Иногда азотная кислота заменяется сухим нитратом в смеси с серной к-той. Последний метод имеет то преимущество, что в технической азотной кислоте содержится значительное количество (до 40%) воды, для связывания которой необходим большой избыток серной кислоты, в то время как при употреблении сухих нитратов эти нежелательные количества воды отпадают. Иногда нитрование ведется оки азота в газообразном виде или в виде нитрозилсерной к-ты. В научном исследовании пользуются также смесью азотной и уксусной к-т, которая играет роль водоотнимающего средства. Весьма энергично и изящно идет нитрование аце-тилнитратом (смешанным ангидридом азотной и уксусной к-т) Ν02· О - СО· СН3. Реакция нитрования протекает в зависимости от следующих факторов: 1) концентрации водоотнимающего средства, 2) темп-ры, 3) состава нитрующего и водоотнимающего агентов.Последние факторы влияют гл. обр. на место введения нитрогруппы; кроме того концентрация водоотиимающего средства и f реакции сказываются на числе введенных нитрогрупп. В большинстве случаев азотной кислоты берут почти теоретич. количество, водоотнимающего же средства—необходимое по расчету (смотрите Нитробензол) для получения оптимальной концентрации отработанной кислоты, и регулируют t° так, чтобы она не превышала оптимальной. Т. к. реакция ни трования экзотермична (так наир., при получении 1 з-моля нитробензола выделяется ок. 36 cal), то ее приходится часто вести при наружном охлаждении, в особенности при получении мононитросоединений. Полинитросоединения получаются при большой концентрации водоотнимающего средства и при более высокой ί°. Роль катализаторов в реакции нитрования не вполне еще выяснена. Известно, что ртуть и ее соединения способствуют не только нитрованию, но и окислению нитруемого тела; так, при нитровании бензола в присутствии ртути получаются динитрофенол и пикриновая к-та. В технике реакция нитрования проводится в специальных аппаратах из кислотоупорного чугуна или алюминия (стойкого к азотной кислоте), снабженных мошной мешй и охлаждающей (нагревающей) рубашкой. Для интенсификации перемешивания и охлаждения предложены нитраторы специальных конструкций. Нитросоединения представляют в большинстве случаев продукты, окрашенные в желтый до оранжевого цвет, и часто обладают специфическим миндальным запахом. Большинство нитросоединений облада-
ет чатыми свойствами.Нитрогруппа качественно, помимо физич. свойств, м. б. определена путем реакции восстановления и по образованию амидной группы при помощи цветной реакции сочетания диазотированно-го амина с R-солью. Количественно же нитрогруппа определяется по Лимпрехту восстановлением точной навески исследуемого нитротела избытком титрованного раствора SnCl2 и обратным оттитрованием непрореагировавшего SnCl2 иодом. Кроме того также применим метод восстановления нитрогруппы титрованным раствором TiCl3 в струе инертного газа.
Нитрование бензола протекает весьма легко; при достаточной концентрации серной кислоты и несколько повышенной (° получается ж-динитробензол, то есть первая нитрогруппа направляет следующие в ж-положение. Необходимо отметить, что введение трех нитрогрупп непосредственным нитрованием бензола не приводит к надлежащим результатам, вследствие чего тринитробензол получается окольным путем из тринитроа—окислением и удалением С02 из трини-тробензойной кислоты по схеме
| сн3 | соон |
| Ν02χλ/Ν02 Ν02χ | 1 /№. |
| и i | J |
| 1
NOs |
1
NO2 |
| нитруется | легче |
N02
,Ν03
NO2
ствие чего может при достаточно крепкой серной к-те и высокой ί° (120—130°) переходить в тринитро. При этом необходимо—во избежание разбавления реакционной смеси водой при предварительном введении двух нитрогрупп—выделять промежуточные моно- и динитропродукты. При нитровании а до мононитропродукта получаются два изомера. Разбавленная азотная к-та нитрует в метильной группе, давая фенилнитрометан. Все стадии нитрования а могут быть выражены сле-дущей схемой
CHjNO.
сн.
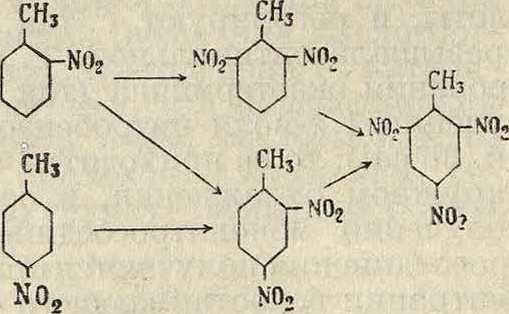
Нафталин в виду особенностей нафталинового ядра нитруется в α-положении по схеме
N0-2
NO2
I
СООО
NOa
СО
i I _
NO2NO2
Получившиеся изомеры динитропродуктов разделяются благодаря значительно большей растворимости изомера 1,8 в органических растворителях. /5-нитронафталин получается лишь окольным путем, исходя из /?-наф-тиламина.
Нитрование антрацена мало применяется, т. к. при этом наряду с присоединением азотной кислоты в мезо-места антрацена имеет место и окисление антрацена.
Антрахинон при действии азотной кислоты переходит в мононитроантрахинон с нитрогруппой в α-месте, который весьма легко нитруется дальше, давая смесь динитроан-трахинонов с преобладанием а, а-изомеров (1,5- и 1,8-)
no.·
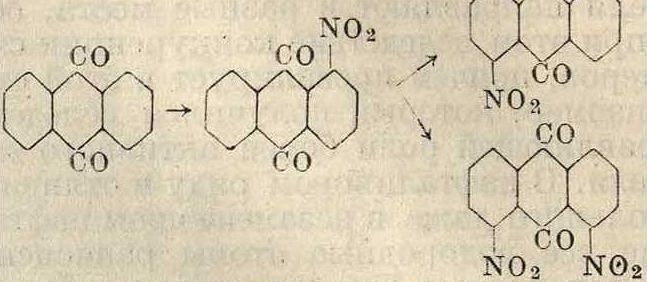
Введение нитрогруппы в /1-место протекает с трудом, но нек-рые производные антрахи-нона, как ализарин, при нитровании в уксуснокислой среде дают 3-нитроализарин, в то время как тот же ализарин при нитровании в сернокислой среде дает 4-нитроализарин. На данном примере видна ориентирующая роль уксусной к-ты.
Фенолы в силу наличия в них активирующей гидроксильной группы нитруются весьма легко. Так, обыкновенный фенол нитруется уже 60%гной азотной к-той в отсутствии водоотнимающих средств в смесь о- и и-ни-трофенолов. Более жесткое нитрование приводит преимущественно к ди- и тринитро-фенолу по схеме он
] N0»
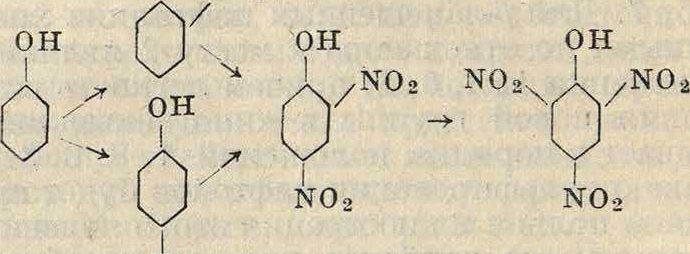
NO2
Те же ди- и тринитрофенолы м. б. получены при каталитич. нитровании бензола в присутствии ртути. Обычно в технике динитрофенол получают не нитрованием фенола, а омылением" динитрохлорбензола, получаемого нитрованием хлорбензола по схеме
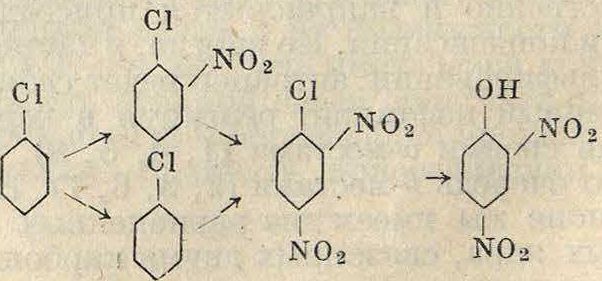
I
N 02
Этот окольный путь представляет то преимущество, что в динитрохлорбензоле хлор весьма подвижен вследствие наличия в молекуле в о- и «-положениях двух отрицательных нитрогрупп, благодаря чему он заменяется, гидроксильной группой при нагревании с водными растворами щелочи, в то время как введение гидроксильной группы до нитрования представляет технически более трудную задачу (смотрите ниже). Кроме того хлорбензол более доступен, чем сульфокислота бензола—
исходный продукт для синтетического получения фенола.
Нафтолы также нитруются весьма легко, причем гидроксильная группа направляется в хиыогенные места. Так, α-нафтол вводит нитрогруппы в положения 2 и 4, то есть в то ядро, где имеется гидроксильная группа, т. к. α-нафтол можно представить как сочетание бензольного и фенольного ядер, а фенол нитруется значительно легче бензола. Но т. к. при этом нитровании всегда имеют место и реакции окисления α-нафтола, то для получения этого ценного динитропродукта (краситель—желтый Марциуса) пользуются сульфированием α-нафтола и заменой подвижных в данном случае сульфогрупп нитрогруппами по схеме
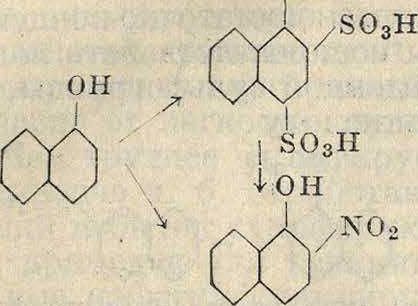
N0.
Аналогично получается и краситель нафтоловый желтый S из трисульфокислоты а-наф-тола по схеме он он
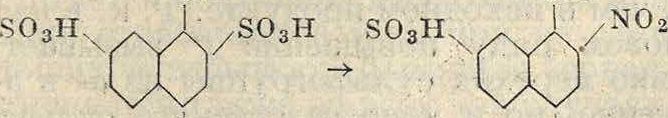
S03H NOa
/5-нафтол -при нитровании также вводит две нитрогруппы в хиногенные места, из которых лишь одно находится в фенольной половине нафтола, а второе в положении 6 (в амфипод ожении, равноценном для /7-производных нафталина—и-положению бензола)
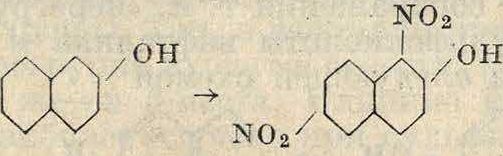
Непосредственное нитрование аминов применяется редко вследствие опасности окислить амины азотной к-той. Для защиты аминогруппы применяется нитрование аминов в растворе концентрированной серной к-ты, но при этом нитрогруппа вступает преимущественно в ж-положение к амидной группе. Так, диметиланилинвэтих условиях дает по схеме
N(CH3)2
, I
N(CH3).H2S04
0 N(CH3b
· I"
N0я.
; ок. 60% ж-изомера и 40% «.-изомера. Для самого анилина эта реакция не применяется, т. к. экономичнее получать ж-нитроанилин частичным восстановлением динитробензола. о- и и-нитроанилины получаются при нитровании защищенного введением ацильной группы анилина и последующим омылением ацильного продукта’по схеме
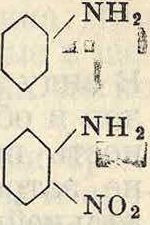
nhcor nh2
i i
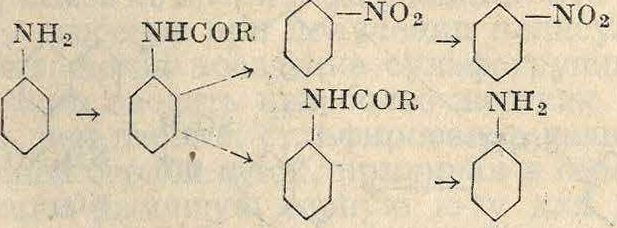
no2 ж>2
Аналогично м. б. получен и 2, 4-динитроанилин. Эти продукты м. б. получены также ами-нированием нитрохлорбензола (смотрите ниже). Соотношение о- и «-изомера при нитровании ациланилинов зависит от характера ацильной группы, от Г нитрования и среды. Однако повышение t° способствует увеличению выхода о-изомера. Точно так же нитрование в уксуснокислой среде или ацетилнитратом приводит почти исключительно ко-изомеру.
Нафтиламины также нитруются в виде ацильных производных, давая 2- и 4-нитро-1-нафтиламины для α-нафтиламина и 1-ни-тро-2-нафтиламин для /5-нафтиламина. Незащищенный /5-нафтиламин нитруется в сильно кислой среде в а-положениях 5 и 8
NHCOR
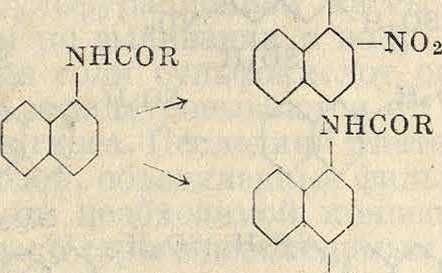
I
N0-2
Х02
I
—NHCOR j/X^-NHCOR
Весьма большое значение в технике имеет нитрование сульфокислот. Для получения нитросульфокислот возможен и путь сульфирования нитропродуктов, т. к. направляющая ориентация нитро- и сульфогруппы одинакова. Но технически в большинстве случаев удобнее нитровать заранее полученные сульфокислоты, т. к. при этом есть возможность, не выливая сульфурационной массы в воду, прибавить к ней необходимое количество азотной к-ты. Оставшаяся после сульфирования серная к-та играет роль водоотнимающего средства. При иной последовательности процессов пришлось бы сначала выделять нитропродукт, сушить его и затем сульфировать дымящей серной к-той при высокой ί°. Эти реакции применяются чаще всего в нафталиновом ряду и служат промежуточной стадией для получения тех сульфокислот α-нафтиламина, которые не м. б. получены непосредственным сульфированием а-наф-тиламина (различная ориентация групп). В приведенных ниже схемах указаны получаемые при этом нитросульфокислоты
OoN so3h
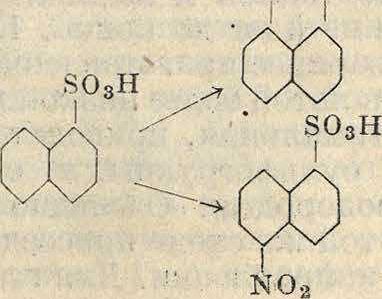

/S03H
no2
no2
00
/SOoH
00
so3H
SO3H I ,Λ-» NOiNOs
NO»
N02
”°* SO3H
Ч
S03H
со
NO>
Как видно из схемы, нитрогруппа преимущественно направляется в то ядро, где нет сульфогруппы, т. к. сульфогруппа затрудняет введение нитрогруппы в свое ядро. Часто нитруются и дисульфо- и трисульфокис-лоты нафталина по схеме
so3h I
NO2SO3H
S03H
303н.
00 ’8О,„/С0
N02S03H
со
so
06 ",чсо
(с трудом)
S03H ^ v xNO SOsH
S03H
SO3H
ч,
/S03H S03H.
S03H S03H
N02
/SOsH
NO 2
S08H4
CO-COw(струдом)
so3H
Ό0
no2
S03H
,S03H S03H4
.S03H
S03H H0js N03
Аналогично нитруются и сульфокислоты антрахинона. Иногда нитрогруппа вводится в уже готовые красители. Интересны опыты по нитрованию оки азота, но исследования в этой области еще не закончены.
2) Реакция сульфирования протекает по схеме II (для бензола), приведенной выше на ст. 163. Для доведения реакции до конца необходимо водоотнимающее средство; в большинстве случаев таковым является избыток сульфирующего агента—серной к-ты. Количество последней берется таким, чтобы по окончании сульфирования оставшаяся (т. н. отработанная) серная к-та имела необходимую, оптимальную концентрацию, которая определяется по легкости сульфирования данного объекта. При концентрации отработанной кислоты ниже оптимальной реакция не идет до конца. Кроме того нагревание заранее приготовленной сульфокислоты с кислотой.более низкой концентрации, чем оптимальная, приводит к так называемым «гидролизу» сульфогруппы, то есть к замене последней водородом. Очевидно эта реакция идет по той же схеме присоединения, но в обратном направлении. Для иллюстрации этого вышеприведенная схема написана в виде обратимой реакции. При нагревании заранее приготовленной сульфокислоты с серной к-той такой концентрации, что исключена возможность как дальнейшего сульфирования, так и гидролиза сульфогруппы, возможны моменты так называемым «странствования сульфогруппы». Это явление особенно распространено в нафталиновом ряду и вообще в тех соединениях, где при сульфировании введение сульфогруппы протекает легко и где легко в силу этого происходит гидролиз сульфогруппы. Приэтом введенная в определенное положение сульфогруппа «перегруппировывается» в другое, более стойкое положение. Так, α-сульфокислота нафталина при нагревании с серной кислотой, недостаточно концентрированной для введения второй сульфогруппы и достаточно концентрированной, чтобы воспрепятствовать полному гидролизу введенной сульфогруппы, переходит в /3-сульфокислоту so3H
со юэ/80,н
Очевидно при этом наступает частичный гидролиз нестойкой, легко введенной сульфогруппы, после чего присоединение элементов серной кислоты идет в ином направлении, и в результате отнятия воды получается сульфокислота с сульфогруппой в ином месте, чем в исходном продукте. Т. к. в нафталиновом- ряду повышение t° вызывает не только переход сульфогруппы из а в ^-положение, но и непосредственное сульфирование в /S-положении, то можно предположить, что при повышенной ί° в нафталине активируется двойная связь между /3-углеродами с возможной осцилляцией двойных связей, в результате чего и присоединение элементов кислоты идет в /3-места и после отнятия воды получается /3-сульфокислота. В силу этих соображений т. н. «перегруппировка» α-сульфокислоты нафталина м. б. изображена следующей схемой
ω+ он к яо3н
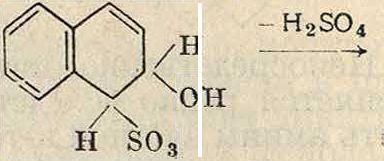
H2S01 ^
ОН + H2SO,
+ I ----1
н
^ч^ч н
h2so4
ч он-н2о н
%/·4 $ο3Η
Соответствующее толкование могут получить и остальные случаи странствования сульфогруппы, о которых будет речь ниже, а также и странствование карбоксильной группы (смотрите ниже). Ход реакции сульфирования зависит от следующих, взаимно связанных факторов: степени легкости сульфирования данного объекта, концентрации и свойств сульфирующего агента, t° реакции и от катализаторов. Степень легкости сульфирования находится в связи с группами, активирующими двойную связь соединения и придающими подвижность водородным его ато мам. Общие закономерности, наблюдаемые во всех реакциях, обусловленных ароматич. характером соединения, находят подтверждение и в реакциях сульфирования. Так, фенолы сульфируются в высшей степени легко. в то время как нитросоединения, сульфокислоты и карбоновые кислоты с карбонильными производными вообще требуют весьма жестких условий сульфирования.
В качестве сульфирующих агентов применяют: серную к-ту различных концентраций, олеум, то есть дымящую серную к-ту, содержащую свободный серный ангидрид, и иногда хлореульфоновую к-ту C1-S02-0H. При применении последней в результате сульфирования образуется и хлорангидрид получаемой сульфокислоты, переходящий в соответствующую сульфокислоту лишь при недостатке сульфирующего агента (от омы-ляющего действия реакционной воды). Выбор концентрации сульфирующей серной кислоты зависит от легкости сульфирования объекта. Чем труднее сульфируется вещество, тем крепче должен быть отработанная к-та и тем больший избыток сульфирующего средства должен быть применен для реакции. Но т. к. значительные избытки сульфирующей кислоты могут затруднить выделение конечной сульфокислоты из сульфурационной массы, то часто сульфирование ведут дымящей серной к-той, свободный ангидрид которой (S03) играет роль водоотнимающего средства; этим достигается получение необходимой концентрации отработанной кислоты при не слишком больших ее избытках. Дымящая серная к-та применяется также во всех тех случаях, когда даже 100 %-ная серная к-та недостаточна для отнятия воды от продукта присоединения. Темп-pa реакции играет двойную роль: е одной стороны, определенная t° является необходимым условием для реакции, т. к. при более низкой t° реакция не идет; с другой стороны, t° реакции влияет на активность двойной связи и на место введения сульфогруппы. Так, в нафталин при низкой t° сульфогруппа входит в а-места, при повышенной же—в /9-места. Влияние катализаторов наиболее изучено при сульфировании антрахинона, к-рый сульфируется при обычных условиях в /9-местах; сульфирование же в присутствии солей ртути протекает в а-по-ложении. Хорошее перемешивание при сульфировании, особенно в моменты загрузки и смешения реагентов, играет существенную роль, т. к. в эти моменты возможны местные перегревы, приводящие не только к нежелательному введению сульфогруппы в иные положения, но и к образованию сульфонов строения R · S02 · R и к окислению сульфируемых продуктов. В технике в силу этого часто пользуются предварительным осторожным растворением сульфируемого вещества в серной к-те и последующим прилива-нием, при строгом соблюдении Р-ных условий, избытка сульфирующего агента.
Для сульфирования применяют чугунные кислотные сульфураторы, снабженные мощной мешй, обогревательными приспособлениями и необходимой арматурой для загрузки реагентов, для взятия проб и для выгрузки. Обогрев сульфураторов производится либо глухим паром," либо огнем, либо по системе Фредеркинга (когда необходима очень равномерная f°). Иногда загрузка суль фирующего агента производится в несколько приемов во время хода реакции. Это имеет место особенно при получении полнсульфо-кислот, когда последние сульфогруппы необходимо вводить при более жестких условиях, чем первые; сульфирование начинают обычной серной к-той, приливая в середине реакции дымящую серную к-ту для дальнейшего полисульфирования. Конец сульфирования определяется по растворимости взятой пробы в воде или в водных щелочах. По окончании реакции вся смесь передавливается в чаны с водой или льдом, где, в случае нерастворимости продукта сульфирования в кислой воде, последний выпадает в осадок и отфильтровывается. Растворимые в воде сульфокислоты м. б. выделены либо высаливанием либо в виде кальциевых солей. Высаливание поваренной или глауберовой солью возможно лишь в тех случаях, когда сульфокислота является более сильной кислотой, чем серная, и вытесняет свободные соляную и серную кислоты из их солей и когда натриевая (иногда калиевая) соль сульфокислоты плохо растворима в кислой воде. Выделение сулыфокислот в виде кальциевых (иногда бариевых) солей состоит в обработке всей сульфурационной массы известью или мелом по выливании в воду. При этом кальциевые соли сульфокислот остаются в растворе и отфильтровываются от выпавшего в осадок гипса. Последний тщательно промывают водой, объединенные фильтраты выпаривают до необходимой концентрации и обрабатывают строго необходимым количеством соды. При этом кальциевые соли сульфокислот переходят в натриевые, раствор которых отфильтровывают от осадка СаС03, так же тщательно промывая последний. Объединенные фильтраты натриевых солей выпаривают либо до начала кристаллизации либо досуха. Изомерные сульфокислоты разделяют дробной кристаллизацией их Na-, К-, Са-, Ва-солей или свободных сульфокислот.
Весьма интересен для техники так называемый бакпроцесс (смотрите). По этому методу кроме сульфаниловой кислоты получают также и две сульфокислоты нафтиламинов: 4-сульфокислоту α-нафтиламина и 6-сульфокислоту /?-нафтйламина.
Так же интересен метод одновременного «сульфирования» и восстановления хинон-ных производных с помощью бисульфита. Так, нитрозо-|8-нафтол при действии бисульфита переходит в 1-амино-2-нафтол-4-суль-фокислоту по схеме
NO NOH nh2
о5/о%со-~с6/ОН
I
so3h
Аналогично индамины и индофенолы (смотрите) при действии бисульфита переходят в сульфокислоты производных дифениламина.
Сульфокислоты, а также их соли представляют в большинстве бесцветные соединения, часто кристаллизующиеся с кристаллизационной водой, в большинстве не имеющие явной <°ил_. Для идентификации их переводят действием РС15 в соответствующие хлорангидриды (т. и. сульфохлориды), которые кристаллизуются из органич. растворителей и имеют явные Рял. Сульфокислоты лafio
S03H
непосредственно применяются в синтезе красителей либо служат для изготовления других П. п.
При сульфировании бензола практически получается моносульфокислота, которая при более жестких условиях (олеум, повышенная 4°) переходит в ди- и трисульфокислоту по схеме
SOsH S03H SO3H
! I I
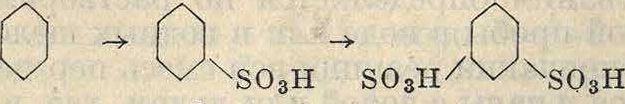
сульфируется чаще всего хлор-сульфоновой к-той, давая при этом два изомера. Один из них—о-сульфохлорид (I)—служит П. п. для синтеза сахарина. Параизомер (II) является отбросом этого процесса и применяется при реакциях этерификации
| СН3 | сн3 |
| /N)4S02C1 | 0
1 S02C1 |
| I. | IX. |
Изомерные ксилолы сульфируются с различной степенью легкости. Легче всех сульфируется м-ксилол, обе метальные группы которого согласовано ориентируют сульфо-группу:
сн3 сн,
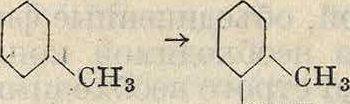
зо3н
Этим пользуются для разделения ксилолов.
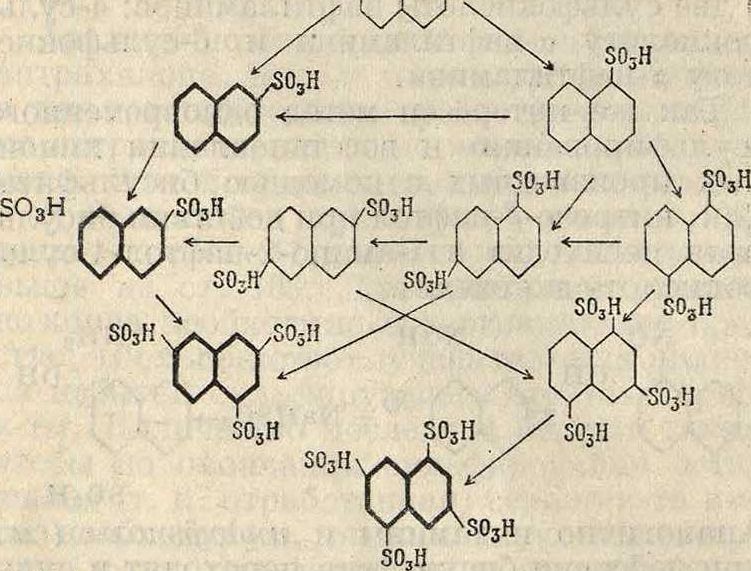
При сульфировании нафталина сказываются особенности нафталинового ядра. В зависимости от 4° сульфирования и от концентрации серной кислоты и олеума получаются различные изомеры. При этом изомеры с более подвижными сульфогруппами могут переходить в более стойкие, гл. обр. /3-изомеры, которые на нижеприведенной схеме изображены более толстыми линиями:
Антрацен при реакции сульфирования обыкновенно дает не моно-, а дисульфокислоты, причем в зависимости от температуры сульфирования получаются либо а- либо /3-диеульфокислоты
so3h so3h so3h

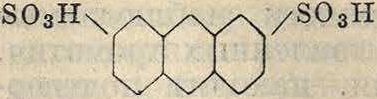
SOsH-7
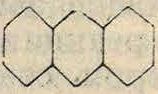
В технике значительно чаще сульфируют не антрацен, а продукт его окисления—ан-трахинон. Сульфокислоты антрахинона являются ценными П. п. для синтеза ряда ан-трахинонных красителей. Антрахинон содержит два вполне равноценных бензольных ядра, которые соединены двумя карбонильными группами, в одинаковой степени затрудняющими сульфирование обоих ядер. Поэтому, несмотря на то что антрахинон сульфируется с трудом, в него входят сразу две сульфогруппы, по одной в каждое ядро. Лишь при сульфировании недостаточным количеством сульфирующего агента (олеума) возможно ввести в антрахинон одну суль-фогруппу; при этом ок. 50% введенного в реакцию антрахинона остается без изменения и регенерируется из сульфурационной массы. Как правило сульфогруппы входят в антрахинон в /3-положение, приводя к следующим дисульфокислотам
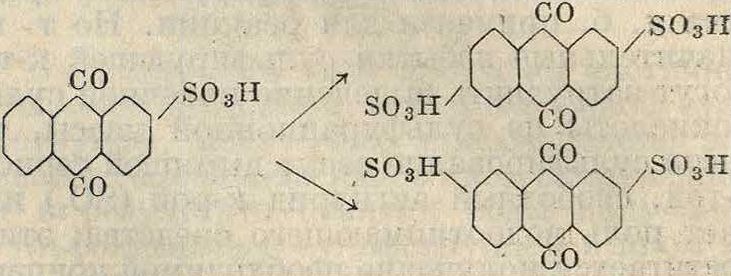
М. А. Ильинским было найдено, что сульфирование антрахинона при тех же условиях, но в присутствии солей ртути, приводит не к /3-, а к а-сульфокислотам антрахинона, причем все сказанное выше о методе получения /3-моносульфокислоты относится также и к способу получения а-моносульфо-кислоты
so3h so3h
I
с°ч
I
/СО
г
so3H
I
чСО
S03H
хсо/к
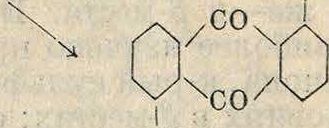
S03II
Интересно, что поваренная соль играет роль антикатализатора и что сульфирование антрахинона в присутствии и солей ртути и поваренной соли приводит вследствие образования и возгонки каломели к /3-сульфокислотам. Комбинируя оба метода, можно получить смешанные"а-/5-дисульфокислоты.
В замещенных углеводородах введение сульфогруппы подчинено общим приведенным выше закономерностям. Легче всего сульфируются фенолы. Обыкновенный фенол сульфируется даже разбавленной серной к-той с содержанием 60—65% H2S04; при этом при пониженной 4° сульфогруппа вступает в первую очередь в о-положение к гидроксилу и лишь при повышенной 4° направляется в и-положение. Сульфогруппы, введенные в молекулу фенола, м. б. легко удалены гидролизом, причем легче гидролизуется сульфогруппа в о-положении. Этим гидролизом пользуются при разделении изомерных крезолов. Схематически указанные превращения выражаются обратимой схемой он
7<
OK
J
V so3h
%
он
S03H
При сульфировании нафтолов сказываются и их фенольные свойства. В первую очередь сульфируется фенольное ядро нафтола; в α-нафтол входят при этом одновременно две сульфогруппы в положения 2 и 4. Хотя положение 2 и является ^-положением нафталина, но в тоже время и о-положением к гидроксилу, вследствие чего легкость введения сульфогруппы в это положение почти равна легкости введения сульфогруппы в положение 4. При жестких условиях эта дисульфокислота принимает еще одну сульфогруппу в положение 7, к-рое является хино-генным (то есть равноценным о- и и-положе-ниям) по отношению к гидроксильной группе и нехиногенным (то есть равноценным ж-по-ложению) к обеим другим сульфогруппам он он он
I
SOsH S08H4
со
/S08H
вании получают весьма ценные дисулвфоки-слоты, называемые по оттенкам, которые они придают азокрасителям,—R (rot) и Gi(gelb), и трисульфокислота по следующей схеме S03H SO3H so3h (СЛ
он
,он
>03Н —I
S03H
/ОН
S03H S03H
/ОН
/ОН SO/И
(R)
S03H S03H ·
Сульфогруппы в положениях 2 и 4 могут он
S03Hf V V so3H
Другие изомерные сульфокислоты /3-нафто-ла получаются соответствующим превращением /S-сульфокислот нафталина или частичным гидроксилированием дисульфокислот (смотрите таблицу в конце статьи).
Сульфирование оксипроизводных антра-хинона производится по правилам сульфирования нафтолов; так, в ализарин можно легко ввести (сульфируя, в мягких условиях) сульфогруппу в положение 3. При более жестких условиях следующая сульфо-группа вводится во второе ядро, а именно в положения 6 и 7 при нормальном сульфировании и в положения 5 и 8 при сульфировании в присутствии солей ртути. Сульфо-группа в положении 3 может быть удалена гидролизом; в этом случае получаются сульфокислоты ализарина по следующей схеме он он
so3h
Ч,
он
1 он
X X
СО ·· SOsH
/С0Ч,
η
S03H
| λ
0 0 |
/γ0Η |
|
0 0 |
^4S03H |
| OH | |
| V
0 0 |
Λ/0Η |
| co | J S03II |
so3H
1 /со,
чсо/
он
so3HN
S03ii S03H
/С0Ч
СО
,он он
/соч(
/ОН
/ОН
ОН
4S03H
/СО
СО
/ОН
sS03H
/соч,
CO
/СОч
-со-7 он
1 он он яо3н легко гидролизоваться, а·также заменяться иными группами, в частности нитрогруппами, чем пользуются для получения динитронафтола и динитронафтолсульфокислоты. Другие сульфокислоты α-нафтола получают окольными путями—превращением сульфокислот α-нафтиламина или частичным гидроксилированием полисульфокислот нафталина (об этом смотрите ниже). /3-нафтол точно так же принимает первую сульфогруппу в фенольное ядро в положение 1. Но суль-фогруппа в этом положении нестойка как вследствие того, что она находится в о-поло-жении к гидроксилу, так и вследствие своего нахождения в α-месте нафталина; поэтому она перегруппировывается в остальные хи-ногенные места.
При сульфировании в сравнительно мягких условиях получается 8-а-сульфокислота /9-нафтола; более высокая ί° приводит к 6-/3-сульфокислоте. При дальнейшем сульфиро-
СО
so3h
/О и
Амины сульфируются несколько труднее фенолов. Кроме описанного выше метода введения сульфогруппы бакпроцессом суль-фогруппа м. б. введена в амины и обычным путем сульфирования. При этом активирование двойной связи и введение сульфогруппы происходят не только в хиногенных по отношению к аминогруппе положениях, но и в нехиногенных, т. к. аминогруппа в сильно кислой среде направляет как кислая группа—NH2-H2S04. Чем выше концентрация олеума, тем больше получается нехиногенных производных. Так, анилин при сульфировании купоросным маслом при 160° даег сульфаниловую к-ту ЗМН2 С6Н, · S03H. Ал-киланилины при сульфировании 25%-ным олеумом дают гл. обр.ж-сульфокислоты. например
N(CH3)2-H,S04 N(CH3)3 h2so4
*
к-рые. служат ценными продуктами для изготовления производных ж-аминофенола.
Наибольшее значение имеет сульфирование аминов нафталинового ряда. Так, а-наф-тиламин дает ряд изомеров по схеме
NHS nh2
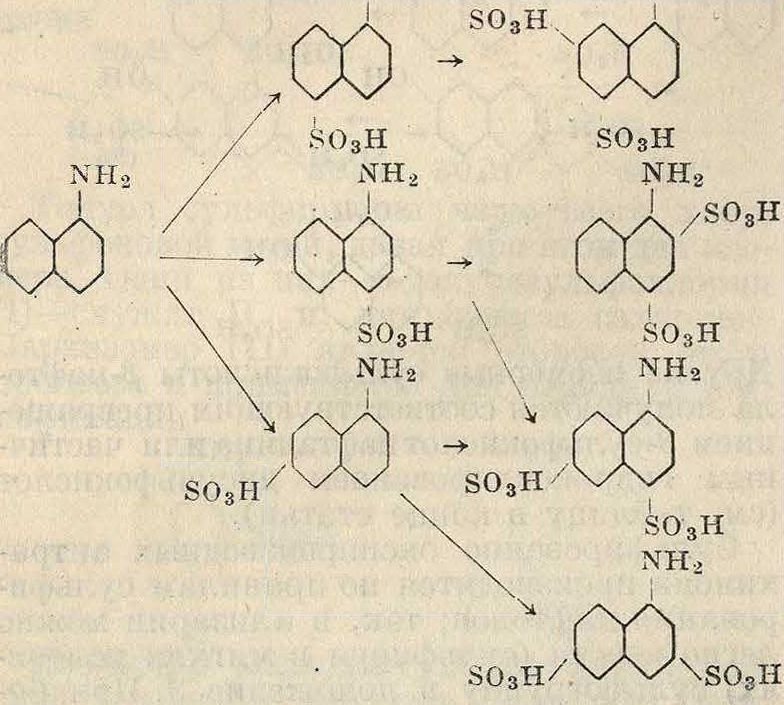
Вследствие этого наиболее ценную 1, 4-наф-тиламинсульфокислоту, называемую н а ф-тионовой, обычно получают запеканием. Другие сульфокислоты α-нафтиламина получают восстановлением нитросульфокислот нафталина или аминированием сульфокислот α-нафтола. /5-нафтиламин сульфируется чаще; его сульфокислоты не могут быть получены восстановлением нитросульфокислот нафталина, т. к. введение нитрогруппы в нафталин в /3-положение прямым путем невозможно. В зависимости от (° сульфирования и концентрации S03 сульфогруппа вступает прочно в а- или ^-положения другого ядра, давая 4 различных изомерных моносульфокислоты, две изомерные дисульфокислоты и одну трисульфокислоту, обладающую легко подвижной сульфогруппой в положении 1, которая возможно является первой промежуточной стадией при сульфировании
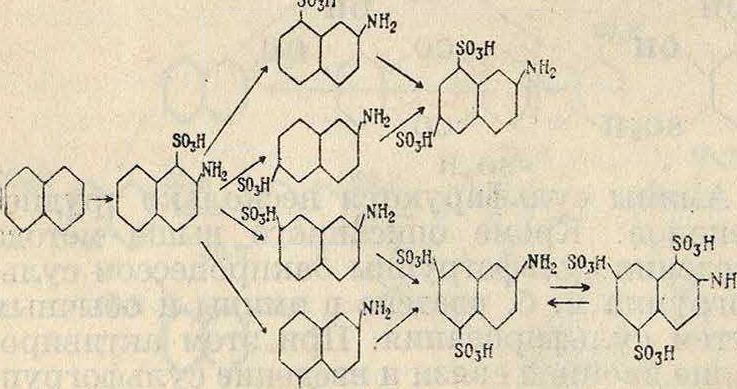
so,я
Дисульфокислоты /З-нафтиламина являются ценными И. п. для изготовления употребительных—в синтезе азокрасителей—сульфокислот аминонафтола: у- и 1-кислоты. Иногда сульфогруппа вводится в готовые красители обычным сульфированием. Число вырабатываемых сульфокислот, особенно нафталинового иантрахинонового рядов, весьма велико.
3) Реакция галоид и рования постепенно приобретает все большее технич. .значение. Из галоидов наибольшее применение имеет хлор. Фтор вследствие неудобств работы с ним не применяется вовсе. Бром и иод имеютвесьмаограниченное применение—
лишь в тех случаях, когда необходимо придать продукту вполне определенные, вызываемые данным галоидом свойства; чаще всего это имеет место у готовых красителей, которые приобретают при введении брома или иода болев ценные качества. Если же галоид вводится лишь временно (для последующей замены его в П. и. другой группой), то применяется почти исключительно дешевый, технически доступный хлор (реакция хлорирования). Реакция хлорирования протекает по схеме III, приведенной на ст. 164. Существование П. п. присоединения при га-лоидировании является доказанным: так, выделены и исследованы продукты присоединения хлора к бензолу и нафталину. Лишь в тех производных бензола и нафталина, где наличие заместителей активирует водород ядра, реакция идет до конца, не останавливаясь на промежуточной стадии. Реакция хлорирования имеет преимущественное значение в рядах бензола и антрахинона, в то время как в ряду нафталина главную роль играет реакция сульфирования. Ход реакции хлорирования (галоидирования) зависит от характера хлорируемого вещества и хлорирующего агента, от f реакции, действия света и наличия катализаторов. Характер хлорируемого вещества сказывается преимущественно во влиянии имеющихся в соединении заместителей, активирующих ход реакции. В качестве хлорирующего агента применяется гл. обр. газообразный хлор, в меньшей мере—хлорноватистая к-та и хлористый сульфурил (S02C12). Вероятное действие последнего состоит в предварительном введении группы S02C1 с последующим удалением S02. В редких случаях применяется хлористая с.ера. Темп-рные условия сказываются главным обр. на разложении П. п. присоединения при повышенной ί°. Этому же разложению способствуют катализаторы, гл. обр. ТеС13 и А1С1Э, расшатывающие систему хлорируемого тела и сообщающие тем самым большую подвижность его атомам. Взаимное влияние i°-Hbix условий света и катализатора будет рассмотрено ниже.
Кроме непосредственного введения галоида реакцией хлорирования существуют окольные методы хлорирования, при которых галоид вводится либо превращением диазосоединений по методу Зандмейера и Гаттер-мана либо превращением сульфокислот при обработке последних хлором in statu nascendi или хлористым сульфурилом (вероятной промежуточной стадией является хлор-ангидрид сульфокислоты); значительно реже хлорпроизводные получаются действием РС13 на фенолы.
При хлорировании бензола всегда образуется продукт присоединения хлора; при достаточном количестве хлора на солнечном свету может получиться и исчерпывающий продукт присоединения—гексахлорциклоге-ксанС6Н6С1«. В присутствии катализатора, гл. обр. ТеС13, из продуктов присоединения выделяется НС1 и получаются хлорзаме-щенные бензола. При подобном каталитич. хлорировании бензола получается не только монохлорбензол, но и дихлориды (иногда и полихлориды), причем количество последних возрастает по мере увеличения содержания в реакционной смеси монохлорбензола и уменьшения бензола. Т. к. полихлориды являются отбросом производства, то обычное хлорирование бензола ведут не до конца, останавливая процесс, когда в реакционной смеси содержится 40—42% С6Н„, 50—55% С6НиС1 и 3—10% полихлоридов. Из этой смеси фракционной перегонкой выделяют бензол и хлорбензол.
При хлорировании а имеет место конкуренция между ароматич. ядром и метальной группой. При наличии катализаторов получаются исключительно хлортолу-олы, в то время как отсутствие катализаторов и действие света вызывают введение галоида в боковую цепь. Повышение Г в отсутствии катализатора также ведет к введению галоида в боковую метальную группу. Сочетанием ряда условий можно добиться получения полихлоридов, содержащих галоид и в ядре и в боковой цепи. Вышесказанное можно объединить в схеме
СН2С1 1
СНС12 СС13I
сн8
сн3
-С1
СН3
сн2С1
I
€1 СН2С!
С1 С1
Нафталин может точно так же дать продукты присоединения, именно нафталинди-хлорид и нафталинтетрахлорид, которые выделяют НС1 даже в отсутствии катализатора (при нагревании). При этом из нафталинди-хлорида получается α-хлорнафталин, а из нафталинтетрахлорида—три изомерных ди-хлорнафталина по схеме
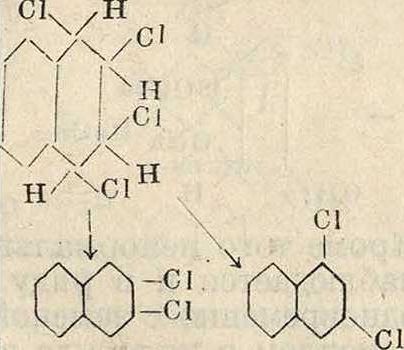
СК /Н
СО<"
I С1 С1
II и
СО 00
С1
При хлорировании расплавленного нафталина в присутствии катализатора получается непосредственноа-хлорнафталин,к-рый дальше хлорируется в тех же условиях уже под влиянием направляющего действия атома хлора по схеме
С1
CI
CI
С1
I
С1
/ϊ-хлорнафталин и другие ди- и полихлорна-фталины получаются" либо из аминов либо чаще из сульфокислот и имеют лишь теоре-
тич. значение, т. к. позволяют идентифицировать различные производные нафталина.
Фенантрен и антрацен присоединяют хлор по своим мезо-местам и далее, отщепляя НС1, переходят в галоидопроизводные по схеме ei. ,н ,сл
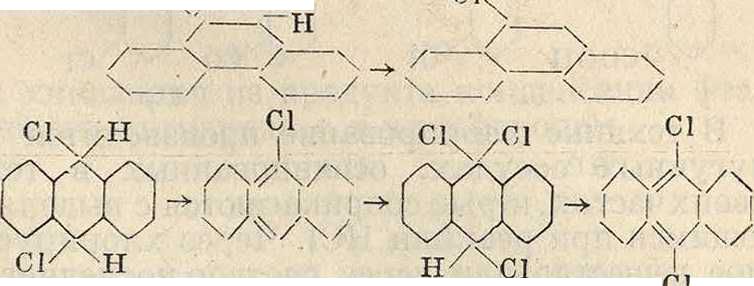
Как видно из этой схемы, по отношению к галоидам в антрацене более податливы места 9 и 10, в то время как сульфирование идет по боковым ядрам.
В антрахиноновом ядре непосредственное хлорирование применяется редко .Чаще хлор-антрахиноны получают из сульфокислот ан-трахинона либо синтезом из фталевого ангидрида и его хлорпроизводных с хлорпро-нзводными бензола.
Фенолы хлорируются весьма легко при действии щелочного раствора хлорноватистой кислоты на фенол по схеме он он
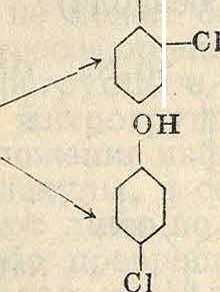
Аналогично α-нафтол хлорируется в смесь о- и w-изомера. Применение хлористого сульфурила приводит почти исключительно к и-изомеру по схеме он он
SO. ‘ НС!
CI
Хлорирование /J-нафтола приводит к 1-хлор-2-нафтолу, к-рый в дальнейшем хлорируется алифатически в кетоформе по схеме Cl н CI С! С1
он Λ×у он Λ V
^00=о Из галоидопроизводных наиболее важны нитрохлорбензолы, в которых влияние нитрогрупп сказывается на подвижности хлора (смотрите выше). И в ряду нафталина продукт нитрования α-хлорнафталина (I)
С1 Ο.Ν С1
III
N02
I.
II.
обладает подвижным хлором, в то время как при хлорировании нитронафталина главным продуктом реакции является изомерный с предыдущим 1,8-хлорнитронафталин (II), в к-ром подвижность хлора меньшая. Оказывается, что лишь в тех галоидопроизводных подвижность хлора велика, в которых в о- и п- (а также равноценных в нафталине)
местах находятся т. н. отрицательные группы. Так например, в соединении (I) хлор значительно подвижнее, чем в получаемом из этого соединения нитрохлорантрахи-ноне (II)
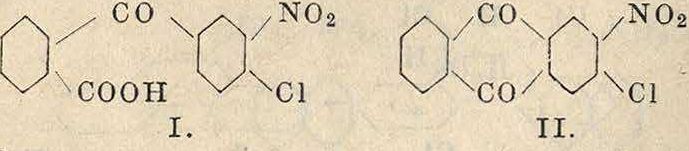
В технике хлорирование производится в чугунных сосудах, освинцованных в тех своих частях, которые соприкасаются с выделяющимся при реакции НС1. Через хлорируемое вещество или через раствор последнего, в инертном растворителе пропускают хлор, регулируя f реакции. Для хлорирования бензола и а предложены периодически и непрерывно действующие колонные аппараты.
4) Реакции гидр оксидирования и а минирования протекают по общей схеме
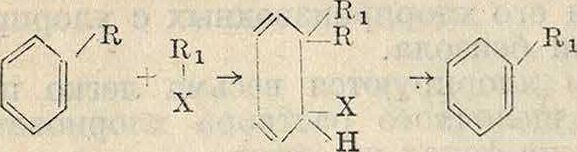
и могут представлять следующие частные случаи (для бензола)
Л
S03Na О Na i
+ Η · "
^Ч |
ONa
SO,Na Η
% τ
NaHS03(Na2S03)
II.
ONa Na0H
+ H *
^4
ONa
C1
/H
4 H
III.
ONa
HCl(NaCI)
,S03Na
I NR
4
/Ri
N
XR2
SO, Na H
H
,R
f. У R;
- NaHSO,[(NH4),SO,!
ΙΛ’.
^ /CI n/Rj r
4C1
/H
II
HCl(NHjCl)
<А/КПз он
-1- I H
y /011
4N1I2/H V4i
,011
+ NHj
VI.
№
,.0H
| /Е, | |
| 4K* | |
| N | 40II |
| " ! R2 | H |
| Η | 4 H |
| n/Ri | |
| j/_ 4R. | H20 |
Гидроксилирование нитропроизводных применяется весьма редко (лишь в антрахиноно-вом ряду) и протекает вероятно по аналогичной схеме. Реакция (I), называемая щ е-лочным плавлением, т. к. технически она проводится путем сплавления солей сульфокислот со щелочами, имеет наибольшее значение и является наиболее удобным методом получения фенолов и их производных. Ее ведут в закрытых чугунных или железных котлах, снабженных мощной скребущей мешй и паровым обогревом. Часто реакция ведется под давлением, когда необходимо вести плавление в водном концентрированном растворе щелочи при t° более высокой, чем t°Kun_ этого раствора. В реакцию щелочного плавления вводят не только сульфокислоты, но и их производные, переходящие в производные фенолов. При щелочном плавлении помимо замены сульфо-группы на гидроксил возможны и побочные явления, как «гидролиз» сульфогруппы, то есть замена ее водородным атомом, или введение вместо сульфогруппы гидроксила не в то же место, а в соседнее. С точки зрения промежу-точных стадий присоединения эти побочные явления м. б. объяснены присоединением элементов щелочи по месту той же двойной связи, но в обратном направлении, по схеме он
yi* ./SO,Na л
-ь !
/Чу 0Ny
он
| TT |
О |
| |XSO,Na | V |
| x ONa — | f |
| /^AH | oh |
NaHSO,
- NaHSO,
Кроме того ненормальное течение процесса наблюдается и в ряду антрахинона, когда одновременно с заменой сульфогруппы гидроксилом в молекуле в о-положение к первому гидроксилу становится второй. Во избежание этого плавление в антрахиноновом ряду (если подобное окисление нежелательно) ведут не с гидратами окисей щелочных металлов, а с гашеной известью. При щелочном плавлении нафталиновых производных особую роль играет подвижность сульфогруппы. Необходимо отметить, что подвижность сульфогруппы при щелочном плавлении не всегда * соответствует подвижности сульфогруппы при ее кислотном гидролизе. При последнем в полном согласии с термо-динамич. положениями наиболее подвижна та сульфогруппа, которая наиболее легко была введена в соединение. При щелочном плавлении мы имеем иные термохимии, условия, в результате которых легче всего уходит сульфогруппа, занимающая по отношению к другому заместителю незаконное место, то есть такое, в к-рое сульфогруппа не могла бы вступить или вступает с большим трудом при не-
посредственном сульфировании. Лишь при прочих равных условиях сульфогруппа в а-положении более подвижна, чем сульфогруппа в (8-положении и при щелочном плавлении. Иллюстрацией служат следующие примеры он * он
so3n
H03s
I
ОНх
S03H
S03H
он но
soall
/ОН
so3H
он
so3H
xso3H so3h
.ОН
-он
Особенностью является также то явление, что сплавление со щелочью ж-дисульфокис-лот и м-оксисульфокислот нафталина приводит не к нафторезорцину, а к о-толуиловой к-те. Очевидно получающийся при этом нафторезорцин под действием щелочи и высокой t° разлагается по схеме он
+ 2Н«0
-он
/СООИ
- CH3.COOH
он»
В технике по этим реакциям получают фенол, Д-нафтол, аминонафтолсульфокислоты Н-,у-и 1-кислоты, оксиантрахиноны и ряд других ценных продуктов, например
S03H nh2 ОН NH·.
I I
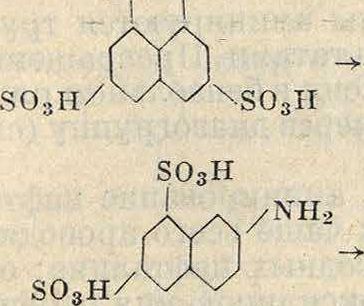
Н-кислота ОН
ЭОзН-7
У·
S03H4/X /NH» SO3H
/NH2
/NH»
I
SO3H ОН
I-кислота
S03H NH. ΟΗΝΗ,
1 1 /SO»H ^4/ix/S03H
S03H SOaH
SS-кислота
OOO OH
/S03H /OH /OH
0 0 0
Реакция (II) называемая иначе омылением галоидопроизводных, имеет преимущественное значение в бензольном ряду и идет наиболее легко при наличии отрицательных групп в п- и о-положениях. В этих условиях ароматически связанный хлор приобретает подвижность, равноценную подвижности алифатич. хлора. Омыление ведут путем нагревания с разбавленными раство рами щелочей. По этой реакции получают например динитрофенол из динитрохлорбензола С1 он
-NO» Jv /NO.2
ч
; I ·
NO;, N02
и хинизарин из продукта конденсации фта-левого ангидрида с «-хлорфенолом он он
/С0Ч
/СО,
СО7 γ -ч7 COCI он
Употребление · вместо щелочи голятов или фенолятов приводит к алкильным или арильным производным фенолов (реакции ксилирования и ароксилирования) ,например
С6Н5ОСН3; 06Ηδ.002Η5; С6Н6-0-С6Н5.
По ур-иям (III) и (IV) в ароматич. ядро вводится амино- или алкаминогруппа. Чаще однако ароматич. первичные амины получают восстановлением нитро- и нитрозосое-динений. Замена сульфогруппы или галоида свободной или замещенной аминогруппой применяется лишь в тех случаях, когда соответствующий амин не может быть получен восстановлением нитросоединения из-затруд-ности введения нитрогруппы в необходимое место. В частности нитрогруппа не может быть введена в //-положение нафталиновой и антрахиноцовой молекулы, и соответствующие амины получают либо по вышеприведенному методу либо превращением окси-производных. Кроме того метод прямого ами-нирования применяется в тех случаях, когда выгоднее ввести замещенную амидную группу одной операцией, например мономе-тил-а-аминоантрахинон наиболее удобно получать по данному методу через сульфокислоту (алкаминированием)
so3H NHCH3
/С0Ч
I
Ш2СН3
/со.
ХС0/Ч^ <Ч^ЧС0/
В то же время //-аминоантрахинон и его производные м. б. получены исключительно ами-нированием /I-сульфокислоты антрахинона, так как //-нитроантрахинон не может получаться непосредственным нитрованием антрахинона. Технически реакция ампнирования проводится нагреванием сульфокислот и ароматич. галоидопроизводных с амидом натрия NaNH2 или, еще чаще, с аммиаком и замещенными аминами. В виду необходимости высоких (° и летучести аминов реакция при употреблении последних проводится в автоклавах под высоким давлением. Из аминов употребляются аммиак, метиламин, этил-амин, диметиламин, диэтиламин, анилин и гомологи последнего. Сказанное выше (при гидроксилировании) о подвижности сульфогруппы и галоида находит подтверждение и при реакции введения амидных групп; так, сульфогруппы в α-положении нафталина и антрахинона могут замещаться аминогруппой при более низкой ί°, чем соответствующие сульфогруппы в /?-положении; точно так же и хлор является более подвижным при наличии в той же молекуле в о- и «-положениях отрицательных групп. Наибольшее значение эта реакция имеет в бензольном и антрахиноновом рядах; в нафталиновом же ряду преимущественное значение имеет реакция замещения гидроксила аминогруппой. В ряду бензола наиболее применимы реакции превращения галоидопроизводных в амины; например нитрохлорбензолы легко превращаются при действии аммиака под давлением в нитроанилины по схемам
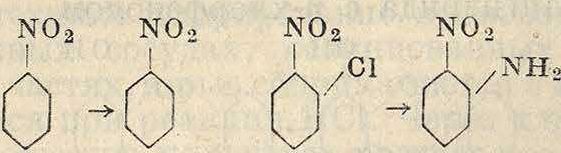
Cl nh2
no2 no2
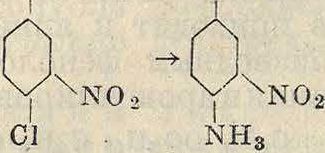
Этот метод получения нитроанилинов является конкурентом обычного метода получения их нитрованием защищенного анилина. Аналогично получаются и нитросульфокислоты аминов по схеме ;
Cl тш2
/SOsH IvSOsH
~>и
i ί I ι·
ΝΟο NOa S03H S03H
При действии на подвижные сульфогруп-пу или хлор ароматич. аминов происходит т. н. а р а м и н и р о в а н и е, то есть введение ариламидных остатков: анилина, толуиди-нов, а- и /3-нафтиламина, аминоантрахино-нов и тому подобное. Высокая t°mm, этих аминов позволяет, при достаточной подвижности суль-фогрупп или хлора, вести эту реакцию без давления. Иногда реакция ведется и с самими аминами по схеме (для дифениламина)
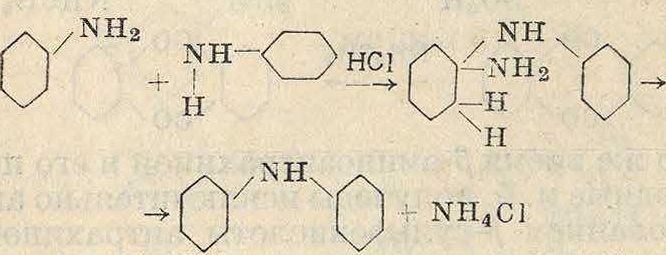
но при этом вследствие значительно меньшей подвижности амидной группы в бензоле приходится вести реакцию в автоклаве под большим давлением. Наиболее легко идет замена хлора ариламидной группой, например
№3—<^>-С! + NH,—Ον
SOnH
-»· N02-O“NH—О
4S03H
I
о
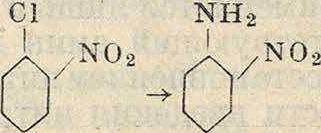
NHCHa
,соч
со7
Br
/со 1 Ann
+ nh2—<^Л-сг
NHCHs
I
NH~0-CH*
Обычно в антрахиноновом ряду, в особен ности при введении остатков аминоантрахи-нона (получение антримидов), требуются катализаторы, например мелко раздробленная медь или соли закиси меди (по Ульману).
В нафталиновом ряду араминирование находит применение по отношению к нафтолам, как и вообще введение любой амидной группы по ур-ию VI. Ур-ия (V) и (VI) м. б. объединены в обратимую реакцию по схеме
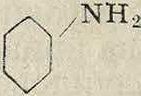
+ н2о
nh2
i i
он н
н
,он
Эта реакция имеет преимущественное значение в нафталиновом ряду. Иногда она применяется и в ряду бензола, гл. обр. для ами-нирования фенолов, в присутствии СаС12или ZnCl2. Таким путем флороглюцин м. б’ легко переведен в аминорезорцин
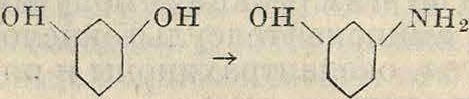
он он
Менее сложные фенолы аминируются труднее и с худшими результатами. Превращение аминогруппы в гидроксил в бензольном ряду-чаще осуществляется через диазогруппу (смотрите Диазосоединения).
Как указано выше, аминирование нафталиновых производных чаще всего проводят, исходя из оксипроизводных нафталина; обратный процесс гидроксилирования первичных аминов нафталинового ряда также находит широкое применение. Эти взаимнопротивоположные реакции проходят особенно легко с помощью найденного Бухерером сульфитного метода, состоящего в предварительной обработке аминов или нафтолов солями сернистой к-ты. Механизм реакции по исследованиям Η. Н. Ворожцова состоит в предварительном присоединении к аминам или нафтолам (реагирующим в кето-форме) элементов сернистокислой соли. При этом наличие у одного кислородного атома двух остатков делает каждый из них более подвижным, вследствие чего его способность к обмену на другую группу, имеющуюся в избытке, возрастает. Эти реакции м. б. изображены следующими схемами он
!
ОН о
S02Na Rx К2
^R! N OS02Na
NaHS03
-СО
СО 00-
Η Η
-ΝΗο * w /ОН
7 -C0 ^00
ΝΗ
/H
Η
Η Η
/os°2Na Η0ΗΧΝΗ, “ *
ω<ϋ-ω
00
Η Η
/OS02Na
NaHSO.
,ΟΗ
+ NaGl -f- SOs
или в виде обратимого процесса по дующей схеме:
ttaHSO
н н
4 /ОН
OSO.Na
юо
еле- i В антрахиноновом ряду часто применяется I замена гидроксила ариламидным остатком—
н н ч н н
00:
он
ΝΗζ
О*
/OSOaNa
nh2
NaHso~
реакция применима и к а- и к /5-производным нафталина; лишь находящаяся в ^-положении сульфогруппа, равно как и сульфогруп-па в о-положении к находящейся в a-положении реакционной группе, препятствует нормальному ходу процесса. Аминирование и гидроксилирование нафтолов и нафтилами-нов м. б. проведены и без участия бисульфита, но при более высоких t° и давлениях, что требует более сложного технич. оборудования; бисульфитная же реакция протекает при нормальном или слегка повышенном давлении и в нек-рых случаях ее можно вести даже в открытых сосудах или-с обратным холодильником. Благодаря этой реакции мы имеем возможность получать целый ряд таких сульфокислот нафтолов и нафтиламинов, получение которых непосредственным сульфированием затруднительно или невозможно, например
so3h
| со/№~ | Q0/OH |
| SOsH | SOsH |
| W- | ->4p0 |
| I
so3H |
S03H |
| он | NHj |
| c0/S0,H’ | сс/80зН |
| so3H | j
so3H |
он
nh2
он
so3H·
-СО - so,„/00
В тех редких случаях, когда бисульфитная реакция не идет, возможно провести гидроксилирование аминов через диазосоединения. В отдельных случаях бывает достаточно одного нагревания амина с разбавленным раствором щелочи под давлением, например
S03H-
&
НО он
SOoH SOoH
<хХ
SOsH
Если при бисульфитной реакции аминирова-ния нафтолов употреблять вместо аммиака его производные, как метиламин, анилин и тому подобное., то легко получаются замещенные амины, например
/γγΝΗ0Η°
со—0
причем реакция идет значительно легче при более низкой t° и с лучшими выходами, чем в отсутствии бисульфита. непосредственным нагреванием смеси реагентов, например он
он
NH-/СО i
чсо/
NH
-о
5) Реакция введения в ядро углеродсодержащей группы протекает по вероятной схеме
с
*Vh
V £
+ нх
В зависимости от характера остатка×реакция проводится либо в присутствии водоотнимающих средств и сопровождается выделением воды из промежуточного продукта присоединения либо в присутствии кислотосвязывающих средств и сопровождается выделением из промежуточного соединения элементов к-ты, чаще всего галоидоводородной. Иногда для удаления последней из сферы реакции достаточно одного нагревания при условии, что реакция проходит в абсолютно безводной среде. Окси- и аминопроизводные обладают и при этих реакциях значительной активностью двойной связи и достаточной подвижностью водородного атома, вследствие чего введение углеродсодержащей группы в их ядро протекает в большинстве случаев при более мягких условиях, невысокой ί° и, чаще всего, в отсутствии катализаторов. Не исключена возможность, что при этом в первую очередь идет образование нестойких продуктов конденсации за счет боковой окси- или аминогруппы, немедленно в тех же условиях перегруппировывающихся в соединения, содержащие углеродную группу в ядре. Нов виду того,что аналогичные реакции идут и с незамещенными углеводородами, приводя к аналогичным продуктам и протекая лишь в более жестких условиях, будет целесообразно рассматривать все эти реакции как непосредственное введение углеродсодержащей группы в ядро. При отсутствии активирующих окси- или аминогрупп, то есть в незамещенных углеводородах реакцию проводят с помощью катализаторов, роль которых еще не вполне ясна; для этого применяется чаще всего безводный А1С1в, реже А1Вг3 и FeCl3; иногда применяют также медь или ее соли. В зависимости от характера углеродсодержащей группы можно получать соединения различных функций: в ядро соединения м. б. введены алкильные и арильные остатки,
различные галоидалкильные группы, карби-нольная, карбонильная, карбоксильная и тому подобное. Иногда здесь возможны вторичные реакции, приводящие к иным, часто более сложным продуктам. В качестве реагирующих с циклич. соединениями веществ применяют галоидгидрины, ы,· альдегиды, кетоны, ароматич. галоидопроизводные и галоидан-гидриды кислот. В качестве галоидгидринов применяются моно- и полигалоидопроизвод-ные, причем в зависимости от условий реакции в результате побочных ее направлений могут получаться производные с различными функциями. При действии на ароматич. углеводороды галоидалкилов в присутствии А1С13 получаются по реакции Фри-деля и Крафтса ароматич. углеводороды с боковой алкильной группой; например этилбензол м. б. получен по схеме
0 +· С2Н5Вг
А1С13
/ч /С2Н3
+ НВг
Применение полигалоидгидринов может при аналогичной реакции в присутствии А1С13 привести к последовательному усложнению продукта реакции; например взаимодействие хлороформа СНС13 с бензолом при наличии избытка последнего приводит последовательно, через ряд реакций конденсаций, ктрифенилметану по схеме
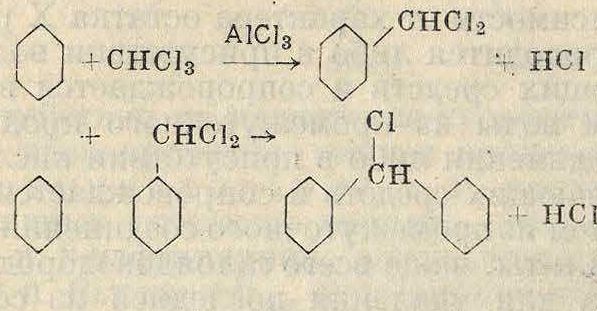
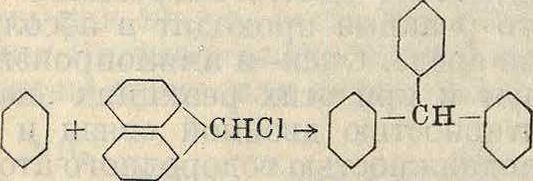
Трифенилметан м. б. получен и минуя первую фазу, исходя из заранее приготовленного хлористого бензилидена С6Н5-СНС12, конденсацией его с бензолом в тех же условиях.
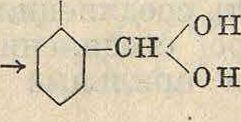
Конденсация того же хлороформа с фенолом протекает при более мягких условиях: для этого достаточно нагреть хлороформ с водным раствором фенолята в присутствии нек-рого избытка щелочи, играющего роль кислотосвязующего средства. Приэтомв первой фазе получается аналогично предыдущему океипроизводное хлористого бензилидена, к-рое в воднощелочном растворе скорее омыляет атомы хлора боковой метильной группы, чем вступает в дальнейшую конденсацию с фенолом. Вследствие этого в результате реакции получается не триокси-трифенилметан, а салициловый альдегид по схеме
ONa
ΓΗ
NaOH сн NaOH
* XCI ”
ONa
Q + CHC18
ONa
ONa
i
0-ch=o
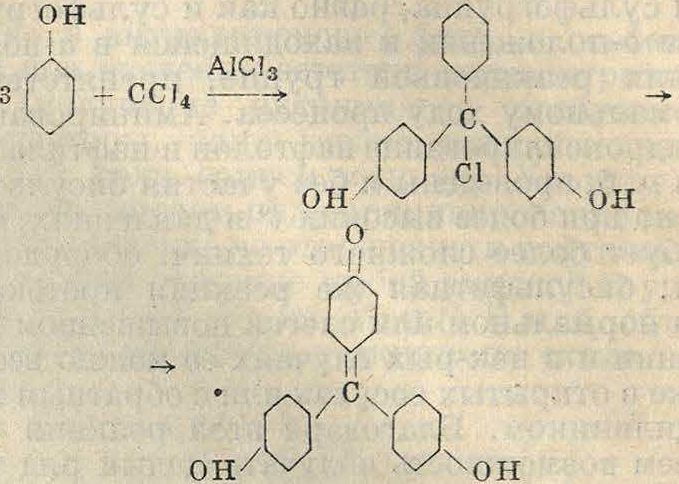
Оксипроизводные трифенилметана получаются при конденсации фенолов с галоидгид-ринами не в воднощелочной среде, а в безводной среде в присутствии А1С13, FeCl3 или ZnCl2. Так например, в результате трех последовательных конденсаций четыреххлористого углерода с 3 молекулами фенола получается при выделении четырех молекул НС1 ро-золовая к-та по схеме он
I
Аналогично галоидгидринам могут реагировать с ароматич. соединениями и кислородные производные; из них доминирующую роль играют альдегиды. Для конденсации последних с углеводородами необходима достаточная подвижность водородных атомов в углеводородах. Альде1 иды при этом реагируют вероятно в ацетальной форме, т. к. реакция идет лучше не с самими альдегидами, а с эфирами соответствующих им ацеталей. Так, нафталин с метилалем СН2(ОСН3)2 дает после двух последовательных конденсаций динафтилметан, вероятнее всего а, а-динаф-тилметан по схеме
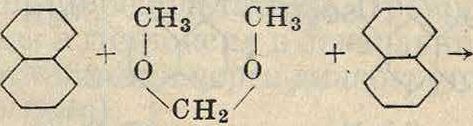
сн2о
У
В присутствии ZnCl2 возможно и иное направление реакции, когда первый продукт конденсации, первичный, под действием ZnClo переходит в соответствующий гало-идгидрин. Так, из бензола и формальдегида м. б. получен хлористый бензил по схеме /СН2ОН ZnCi20/CH2ci
С фенолами и аминами конденсация альдегидов идет значительно легче. При этом в зависимости от условий реакции могут получаться продукты большей или меньшей сложности. При конденсации фенола с водным раствором формальдегида получается в первую очередь и-оксибензиловый, переходящий при избытке фенола и более жестких условиях в диоксидифенилметан
,сн,он он сн2
ОН
он он он
/СН2ч
у
ОН
Специфические условия конденсации приводят к более сложным, нерастворимым продуктам конденсации, называемым бакелита-
ми (смотрите) и применяемым в качестве искусственных смол.
Аналогично и амины легко конденсируются с альдегидами, причем первичные амины дают при этом азометины (смотрите ниже). Третичные амины аналогично фенолам вводят углеродсодержащую группу в ядро, давая ди- и трифенилметановые производные. Так, диметиланилин с бензальдегидом дает после двух последовательных конденсаций тетра-метилдиаминотрифенилметан
<CH3),N
ЧМ(СН3)2
сн=о
/сн
(CHsJsN7 v ν nN(CH3)2
Конденсация ароматич. соединений с карбоновыми к-тами или их галоидангидридами приводит к различным, в зависимости от взятой к-ты, карбонильнымпроизводным,властности к альдегидам и кетонам. Иногда нестойкие галоидангидриды к-т, например хлоран-гидрид муравьиной к-ты, реагируют in statu nascendi. Так, из бензола и газообразной смеси СО и HCi, которая в присутствии А1Вг3 реагирует подобно хлористому формилу НСОС1 (в свободном состоянии неизвестному), получают с достаточными выходами бензаль-дегид по схеме
А
с-о -
ХС1
,0-0
НС!
СО в этой реакции м. б. заменена синильной к-той, реагирующей в изоформе и дающей имид альдегида, переходящий в тех же условиях в альдегид
СН NH
сн=о
/Н
+ C-NH
ЧС1
Если вместо производных муравьиной кислоты в реакцию введены производные уксусной или более сложных к-т, то в результате реакции получаются не альдегиды, а соответствующие кетоны. Так, из бензола и хлористого ацетила получается в присутствии А1С13 ацетофенон
,со·сн3
cico - сн3 -> I I + НС1
Соответственно из а (дихлорангидри-да угольной к-ты) и бензола в присутствии хлористого алюминия получается путем двух последовательных конденсаций бензофенон хх /С0
I + CIC0C1
При замене а в этой реакции его моноамидом получается в результате одной конденсации бензамид
. .co-nh»
A1C13 /Ч
+ ClCONHj------>( J + НС1
Наибольшее значение имеет_ конденсация углеводородов с ароматич. карбоновыми кис лотами, приводящая к производным бензоил-бензойной к-ты
,со · свн3
чсоон
Последние путем замыкания цикла переводятся в производные антрахинона, благодаря чему можно получить целый ряд производных антрахинона, равно как и сам антра-хинон, синтетич. путем. Наиболее употребителен метод конденсации углеводородов (бензол, нафталин и тому подобное.) с фтале-вым ангидридом по схеме (для а)
СО
сн;
f
о
/СО 0_
—н со
СН;
/С0Ч
-о-с-
А1С1.
ч н
А1С13-->
сн^ соон Иначе производные бензоилбеызойной кислоты м. б. получены при конденсации производных 2 молекул монокарбоновой к-ты, например резорцинкарбоновой кислоты по схеме он I
он
/ЧА
соон он
I
носочЛ/он H2S04 А/С°чхч
ОН7
/Ч/Чсо
он он он он
С фенолами конденсация карбоновых к-т и их галоидангидридов идет значительно легче: иногда даже отпадает необходимость применения более реакциеспособных галоидангидридов кислот. Так, фенолы дают легко при действии карбоновых κ-τηΖπ02 окси-производные кетонов. Напр. резорцин при этих условиях дает резацетофенон и резо-бензофенон по схеме (для последнего) он
+ ноос
-о
011
ZnCl2
ОН
I
OH/0/C°O
+ н20
Аналогично /Чнафтол дает а-ацето-/?-на-фтол по схеме со · сн3
он
+ сн3·соон
I—он
+ Н20
С галоидангидридами кислот реакция идет в высшей степени легко. Так, диметиланилин, реагируя последовательно в две стадии двумя молекулами, с одной молекулой а дает в конечном итоге т.н. кетон М и х-л е р а—тетраметил-и-диаминодифенилкетон, ценный П. п. для синтеза ряда основных красителей
ei C1
;/0*со + 0 -
(CH3)2N
4N(CH3)2
(CH3)aN/- v nN(CH3)2
Эта реакция протекает при пропускании а через диметиланилин, причем избыток послед iero играет роль кислотосвязывающего средства.
Точно так же при получении производных бензоилбензойной кислоты наличие гидроксильных гр пп в исходном продукте облегчает течение конденсации с фталевым ангидридом и позволяет вести реакцию без катализатора, лишь в присутствии серной к-ты. Применяемая при этом борная к-та преимущественно этерифицирует гидроксильные группы. При этой конденсации сразу получают производные антрахинона. Так, гидрохинон с фталевым ангидридом дает при такой конденсации 1,4-диоксиантрахинон (хинизарин) он со.
-2НС1
о<
,00—,
хсо-
он он
H3S04
он
h2so4
όοοάοο
он со I
он
I
он
Другие карбоновые к-ты, реагируя с фенолами в присутствии ZnCl2, дают иные продукты конденсации по схеме он.
0=С—R
и онч
<
ХР
он онгЛ
J/U
он
Эти продукты представляют уже не П. п., а основания метиленхинонных красителей (смотрите Красящие вещества синтетические).И сама угольная к-та также может служить для введения углеродсодержащей группы в ядро соединения. Так, при пропускании С02через бензол в присутствии А1С13 получается нек-рое количество бензойной к-ты:
11+ со,А1Сз соон
О
V H
С фенолами эта реакция идет в высшей степени легко. Так, по синтезу Кольбе получается в результате перегруппировки эфира салициловая к-та по схеме
ONa I
,0—GOONa
СО
ONa ONa
ОН
COONa l
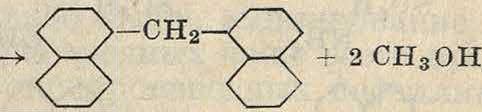
Аналогично Днафтол при той же конденсации дает две оксикарбоновые к-ты, из которых первая получается при 120—145° и переходит при повышении ί° в более стойкую 2, З-нафтолкарбоновую к-ту соон
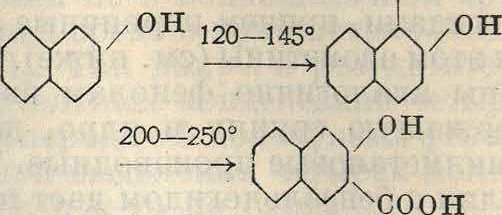
Аналогично предыдущему в ядро соединения м. б. введен и ароматич. остаток. Напр. при взаимодействии бензола и бромбензола в присутствии А1С1а м. б. получен дифенил
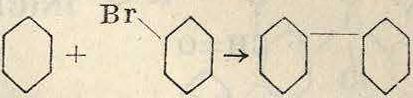
Аналогичные диарилы м. б. получены при конденсации двух молекул галоидопроизводных в присутствии Ζη или Си. Так например, при действии мелко раздробленной меди на α-хлорантрахинон получается 1, 1-диантрахиноиил о ei о о
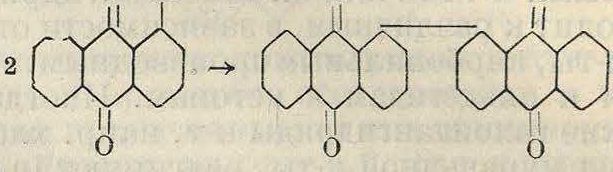
Б. Реакции, обусловленные свойствами боковой цепи соединения. В подавляющем большинстве эти реакции возможны как для алифатических, так и для ароматич. соединений, и для проведения их достаточно лишь наличия активных групп. В ароматич. ряду эти реакции часто облегчаются благодаря влиянию ароматич. ядра, но иногда наличие последнего вызывает и побочные реакции вторичного· порядка. Как общее правило можно отметить, что в результате реакций этого класса органоген, связанный с ядром соединения в исходном П. п., остается в той же связи и после реакции конденсации, которая имеет место лишь в боковой ветви соединения. Среди реакций этого типа наибольшее применение имеют следующие: 1) введение ацильного остатка в аминогруппу или в гидроксил; 2) введение алкильного остатка в аминогруппу, гидроксил или метальную группу; 3) введение гидроксила или аминогруппы в метальную группу; 4) введение метиленового (=СН2) или имидного (=ΝΗ) остатков в ами-ΗΟ-, метил-, нитрозо- и альдегидную группы.
1) Реакция ацилирования (введения остатка к-ты) имеет либо самостоятельное значение либо применяется в качестве временной меры защиты П. п. при нек-рых операциях. Так, при нитровании аминов, для защиты их от окисления за счет азотной кислоты вследствие реакциеспособности аминогруппы, последнюю ацилируют, вследствие чего амин становится более стойким. По окончании нитрования и выделения нитропродукта аминогруппа м. б. освобождена от защищавшей ее ацильной группы реакцией дез ацилирования или омыления, в результате чего получаются нитроарилами-ны. Точно так же при окислении аминопроизводных гомологов бензола в соответствующие аминокарбоновые кислоты амидная группа защищается ацилированием от действия окислителя. В качестве ацилирующих агентов применяют кислоты, их ангидриды и га-
лоидангидриды, вероятно дающие промежуточные продукты присоединения. Реакция ацилирования протекает по след, схемам
I. RNH2 + OHAc-
II. RNHi + O—(Ас)а·
/Ас
-R—N-OH-I н
Н
,Ас
>RN<
VO Η Η
• R-NHAc + Η20
-Ac
• R-NHAc+AcOH
III. RNHa+Cl—Ac -
.Ac
,Π—n -> R-NH-AC4-HCI
/VC1 H H
где Ac—ацильный остаток. По схеме (I) ацилирование идет с выделением воды, которая связывается избытком ацилирующей кислоты или удаляется из сферы реакции нагреванием. По схеме (II) ацилирование идет весьма легко, т. к. в процессе реакции получается не вода, а безводная к-та, которая паевою очередь обладает ацилирующими свойствами. Для более спокойного течения процесса в этом случае иногда рекомендуется вести реакцию в среде инертного растворителя, как бензол, или ксилол. По схеме (III) реакция сопровождается выделением галоидоводородной к-ты, для связывания которой можно применять безводную натриевую соль той к-ты, хлорангидрид которой взят для ацилирования. Иногда одновременно кислотосвязывающим средством и растворителем служит пиридин или другое органич. основание.
Ацилирование аминов можно рассматривать так же как амидирование кислот. Так, бензоилирование анилина является в то же время фениламидированием бензойной к-ты
/nh2 соош +
O0”H~c°O
/ОН ОН—СОх^
Ацилирование же фенолов м. б. трактовано как получение сложных эфиров третичного а (фенола) и кислоты. Так, соответствующий бензоиланилину бензоилфенол является фенильным эфиром бензойной к-ты
Χ>0/Ο~“Ό
В качестве ацилирующих к-т в технике применяются карбоновые кислоты (угольная, муравьиная, уксусная, щавелевая, различные ароматич. к-ты) и минеральные к-ты, наир, серная. Иногда применяют ароматич. сульфокислоты, преимущественно в виде их хлор-ангидридов. Продукты ацилирования называются арилидами с соответственным латинским названием кислоты, играющей роль ацилирующего средства. Так, производные муравьиной кислоты называютсяф о р-м арилидами, производные уксусной кислоты—а цетар и лидами, производные щавелевой к-ты—о ксаларилидами. При действии двуосновных к-т, как щавелевая к-та, возможны диарилиды и моно арил иды, называемые арилокс-аминовыми к-т а ми. В зависимости от взятого амина название «арилид» заменяют корнем названия исходного амина, например
/Н /СН3
0-C=O 00-NH-C=O
I. II.
/NH—СО—СО—NH
СН3 СН;
III.
NH—СО—СООН
СН;
IV.
соответствуют нижеследующим названиям: I—ф орм анилид, II—а ц е τ-β-н афта-лид, III—о к с а л-о-т о л у и д, IV—п-то-лилоксаминовая кислота. Обычш-простейшие арилиды получаются при нагревании исходного амина с соответствующей к-той, причем характер кислоты играет роль при дальнейших реакциях анилида.
Из анилидов в технике особенно важен ацетанилид, получаемый нагреванием анилина с уксусной к-той, лучше всего в алюминиевых ацетиляторах. Для этой операции нет необходимости брать ледяную уксусную к-ту, т. к. реакция идет с 80%-ной к-той, но в процессе реакции приходится отгонять выделяющуюся при реакции воду вместе с частью к-ты, получая в отгоне в среднем 30%-ную к-ту. Все же выход анилида стоит в связи с концентрацией ацилирующей к-ты. В производстве П. п. ацетанилид является промежуточной стадией при получении нитроанилинов по общей схеме
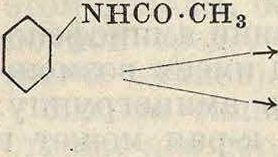
/NHCO-СНз
I
N02
/NHCO-CHs
no2
Значительно труднее ацилируются вторичные ароматич. амины. Так, дифениламин ацилируется лишь при действии^ уксусного ангидрида
.со—сн:+ о<
ХСО-СН;
При действии уксусного ангидрида наами-нодифениламин ацилируется 1-ю очередь его первичная аминогруппа
^-nh-^-nh^Q7 х0
I
NHCO СНз причем эта реакция идет лучше всего в инертном растворителе (бензол). В качестве ацилирующих агентов применяются также ароматич. карбоновые к-ты—бензойная, салициловая, нафтойная и оксинафтойная.
Наибольшее технич. значение имеют арилиды 2-нафтол-З-карбоновой к-ты, или β-оксинафтойной к-ты, являющиеся ценными П. п. для получения на волокне ледяных азокрасителей и выпускаемые в продажу под названием нафтолов AS (смотрите Нафтолы). Простейший из них—анилид /9-оксинафтойной к-ты—получается при обработке анилина нафтолкарбоновой к-той в присутствии РС13 и нейтрального растворителя (а)
0-”н-“;00
Более сложным представителем является нафтол AS марки BR—дианизидид /ί-оксина-фтойной к-ты
,С0— NH Л >. /NH — соч
т
чон осн3х v ^ хосн3 он7 При действии на амины серной к-ты, а также ее хлорангидрида и хлорангидридов ароматич. сульфокислот получаются либо сульф-аминовые кислоты либо арилсульфоариламиды. Сульфаминовые кислоты являются иногда промежуточными стадиями при сульфировании аминов. Из хлорангидридов ароматич. сульфокислот наиболее применим и-суль-фохлорид (отброс сахаринового производства), дающий, наир, с анилином, и--сул ьфофениламид
ci-so2
чсн3,νη—so2·
Ч,
/NH.
CH3
Фенолы ацилируются значительно труднее аминов. Лучшие результаты получаются при действии на фенол галоидангидрида кислоты в присутствии кислотосвязывающего средства, как пиридин и тому подобное., или при действии того же галоидангидрида на фенолят натрия о Na
C1COR-
ocoR
В виду этого при ацилировании аминофено-лов в обычных условиях мы имеем возможность проацилировать лишь аминогруппу, не затрагивая оксигруппы, которая может в дальнейшем подвергаться любым операциям, в которых наличие свободной аминогруппы препятствовало бы желательному течению процесса. Так, при получении алкилированного в оксигруппе и-аминофенола возможно заранее защитить аминогруппу ацилированием, удалив по окончании реакции ацильную группу омылением
/NHa ^ /NHCO-СН3
ОН
ΟΗΧ
/NHCOCHs
/О
/NH3
ОДВО7 v с2н5о/
Ацилирование о-аминофенолов и о-диами-нов обычно не приводит к истинным продуктам ацилирования, так как последние, легко выделяя элемента.! воды или кислоты, замыкаются в циклы типа оксазола или имидазола по следующим схемам /NHa
+ сн.соон -»
Ν/ΧΟΗ
/NH4
ч он
II.
СО-СНз
/NH;
xNH2
/N.
хс-сн3 : Н.О
/NH4
COCH3
Ч-νγττ
СО-СНз
0
СНз-СООН-
,/Ν·
ССН3 + СН3СООН 4NH/ ~ v SNH/
Ациларилиды и ацилированные фенолы являются довольно стойкими веществами и лишь при длительном нагревании с разбавленными щелочами и разбавленными к-тами дезацилируются или омыляются, переходя в соответствующие амины и фенолы по схемам
R-NH CORi;+H20^-R NH2 + R.iCOOH,
R-O CORi + HaO -> R OH + RiCOOH.
2) Реакция алкилирования, т.е. введения алкильного остатка, идет по общим правилам, установленным Гофманом и для жирных аминов, и имеет в ароматич. ряду при производстве П. п. весьма большое значение, т. к. алкилированные амины и фенолы обладают иными свойствами, чем неалкилированные исходные продукты; кроме того красящие вещества, получаемые из алкилированных аминов, обладают более ценными качествами и более глубоким цветом, чем аналогичные красители из неалкилированных аминов. Алкильная группа м. б. введена в тиофенолы, амины, фенолы и—каталитически—в углеводороды с боковой метильной. группой. Наиболее легко алкилируется SH-, затем NH2- и наиболее трудно ОН-группа; СН3-группа вовсе не алкилируется при обычных условиях. Вероятнее всего легкость алкилирования зависит от силы остаточного сродства органогена и подвижности водородных атомов при нем. При этом вероятны промежуточные продукты присоединения. Общая схема алкилирования следующая
RSH + СН8×-► RSCH3 4- ХН
RNHa + СН3Х -> RNHCH3 + ХН
ROH + СН3×-4 R-0-CH3 + ХН
RCH3 + СН3×-> RCHs-СНз + ХН
Алкилирующими средствами являются ы, галоидопроизводные и алкильные эфиры серной кислоты (диалкилсульфаты и алкилсер-ные к-ты), а также соответствующие производные ароматических сульфокислот, например и-сульфокислота, причем наиболее сильным алкилирующим средством является диалкилсульфат.
В технике наибольшее значение имеет алкилирование аминов, в частности анилина, проводимое при действии соответствующего а на анилин при высоких ί° и давлении в присутствии небольшого количества соляной или серной к-ты. При этом возможна промежуточная стадия образования галоидопроизводного или эфира серной к-ты, которые фактически и алкилируют. В результате процесса получается при избытке алкилирующего агента диалкиланилин, например диметиланилин по схеме
+ 2 СН3ОН ->
N(CH3)2
+ н2о
Реакция получения диметиланилина идет в две стадии, то есть алкилирование протекает по ступеням по схеме
Г J + СН3Х^[ J + нх.
/NHCHs
/N(CH3)2
СН3Х -
нх
Скорость алкилирования в обеих фазах примерно одинакова. Поэтому при взаимодействии анилина с одной молекулой метилового а в результате алкилирования получается не исключительно монометиланилин,
а смесь его с анилином и диметиланилином, разделение которых довольно затруднительно.
Аналогичноидеталкилированием в остальных случаях. Применение галоидалкилов не меняет общего хода процесса, но ускоряет и облегчает его течение. Ценные в качестве П. п. и для военных целей простейшие мо-ноалкиланилины, получаемые при обычном алкилировании в смеси с первичными и третичными аминами, м. б. получены окольными путями. Для этого можно временно заменять один из водородных атомов аминогруппы ацильным остатком, к-рый по миновании надобности убирается, либо получать моноалкиланилин элиминированием С02 из алкильного производного, содержащего в боковой углеродсодержащей цепи карбоксильную группу, либо получать моноалкиланилипы восстановлением т. наз. азометинов, или шиффовых оснований
/NHa rN/NH-S02-<^)-CH3I.
, NHCH3
СН3
,Ν-SOa—<(^>—СН3
II.
III.
/NH2 CICH2 C00H
/nhch2cooh_
co2
,nh2
•0
неон
-NHCHa
/N r:CH2 +H
,NHCH3
Кроме собственно алкильного остатка в аминогруппу м. б. введены и более сложные остатки, например —СН2-СООН,—СН2—( )> и тому подобное. Введением остатка уксусной кислоты получают производные гликоколя (глицина)— ценные продукты, служащие П. п. для синтеза индигоидных красителей. Так, при действии избытка анилина на монохлоруксус-ную к-ту (I) получаем фенилглицин (смотрите Индиго). Аналогично при конденсации анилина с хлоргидрином этиленгликоля (II) получается ω-оксэтиланилин
nh2
+ CICH2-COOH-
,νη2· HCl
II.
,nh2
,NH2-HC1
,ΝΗ-СНг COOH
CICH2-CH20H -
NH—CHs-CHtOH
В аналогичную конденсацию вступает также и о-карбоновая к-та анилина—антраниловая кислота (III) и ее содержащий серу аналог— о-тиофенолкарбоновая к-та (IV), которые вводятся в реакцию в виде натриевых солей
A/NHa
ш· о
4COONa
,SH
+ ClCH2 COONa
4COONa
/NHCH2COONa
чсоон
,S-CH2-COONa
+ NaCl
COOH
Соответствующие фенилглицины могут быть получены и из азометинов—присоединением элементов синильной кислоты и омылением получаемого нитрила по схеме
NH-CHa-CN
/NH—СН2—СО ОН
Арилалкильный, преимущественно бен-зильный остаток—СН2—<^>, вводится весьма легко при действии хлористого бензила С1СН2—ζ у или, еще легче, при обработке соединения лейкотропом (смотрите), представляющим хлористый диметилбензилфениламмо-ний строения
СНз сн3
rf-CH.-O
Cl
бензилирующийкакбы in statunascendi,причем бензилирование идет в щелочной среде при непрерывной отгонке острым паром выделяющегося при этом днметиланилина. При этом фенолы, более реакциеспособные в щелочной среде, бензилируются очень легко (легче аминов).
Весьма сильными алкилирующими средствами являются эфиры серной кислоты и аро-матич. сульфокислот, применяемые гл. обр. при алкилировании фенолов, которые вообще алкилируются труднее, чем амины. Для этого Служат главн. обр. алкил сульфаты (I), алкилсерные кислоты (II) и алкильные эфиры и-сульфокислоты (111), получаемые из отброса сахаринового производства—и-то-луолсульфохлорида
so
OR
so
OR
OHs-O-SO.O-R
N0-R 4>H
I. II. III.
Применение алкилсульфатов позволяет вести реакцию алкилирования без давления, но при этом используется лишь половина алкильных остатков, т. к. алкилсульфаты переходят в алкилсерную к-ту, которая сама способна алкилировать лишь под давлением.
Сочетая различные методы алкилирования, возможно получать смешанные алкильные производные аминов и аминофенолов. Так, получив моноалкиланилин, можно ввести в него бензильную группу и получить ценные П.п.,например этилбензиланилин
/С2н5 ——J
Г"Чсн.гО
Соответственно можно получать смешанные алкильные производные аминофенолов, например диметиламин-и-фенетол
~/СНз Л7" хсн3
Наиболее трудно протекает алкилирование метальной группы в боковой цепи соеди-
нения; для этого требуются сильно действующие реагенты или катализаторы. Реакция вполне аналогична синтезу жирных углеводородов по Вюрцу, наир, дифенилэтан получается при действии цинка или натрия на две молекулы хлористого бензила
2 <3-СН2С1 + 2Na — 2NaCl +
+ <^-сн2-сн2-0
Следующий класс реакций, обусловленных характером боковой цепи, состоит в типично жирном омылении галоидопроизвод-ных, содержащих галоид в боковой углеродсодержащей цепи; например при омылении хлористого бензила (I), хлористого бензи-лидена (II) и бензотрихлорида (III) получаются соответственно бензиновый, бен-зальдегид и бензойная кислота
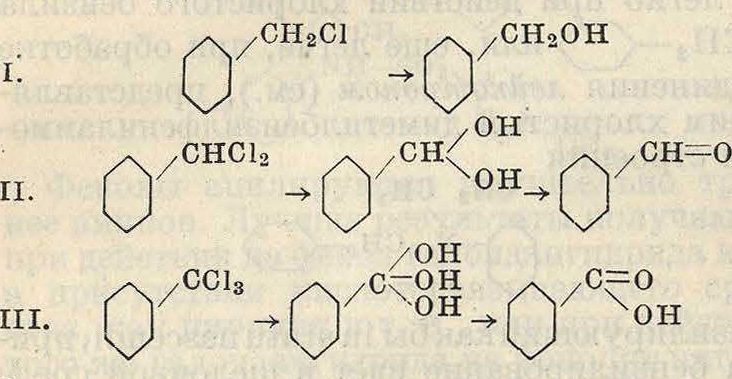
Эти реакции проходят довольно легко при действии щелочей и даже известкового молока. С этими способами получения ароматич. альдегидов и карбоновых к-т конкурирует способ окисления гомологов бензола (смотрите ниже, а также Окисление).
Наряду с заменой атома хлора гидроксильной группой возможна замена его и аминогруппой. Фактически реакция бензилирова-ния анилина является в то же время и фенил-аминированием хлористого бензила по следующей схеме
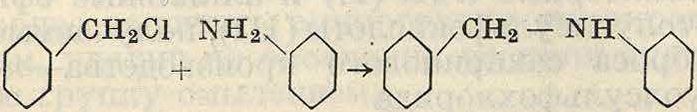
3) Реакция введения в боковую цепь двувалентной свободной или замещенной метиленовой или и м и д н о и группы имеет весьма большое значение при производстве П. п. Эта реакция в большинстве случаев ведется в разбавленных растворах, сопровождается выделением воды, и для проведения ее необходимо наличие в двух реагирующих веществах реакциеспособных групп двух типов: одна из групп должна обладать двумя подвижными атомами водорода, другая же — подвижным атомом кислорода, связанным двойной связью с углеродом или с азотом. Такими группами являются: с одной стороны, метил (в особых условиях) или аминогруппа, а с другой,—карбонил (преимущественно в альдегидах) или нитрозогруппа. Вероятнее всего эти реакции протекают отнятием воды из промежуточного соединения и м. б. выражены в след, схемах
/Ri /Ri
(A) R-CHS + О : С< R-CH2 C(OH) ->
XR2 xR2
/Ri
->R-CH=C + H20
XR2
(Б) R-CHS + О : N-Ri -* R-CH2-N(OH)Ri -»
->R CH : N-Ri + H20
(B) RNHj + О : С/Й‘ ^ RNH-C(Oh/Ri ->
XR2 xR2
-> RN : c *+H20 4R2
(Г) RNH2 + О : N Rx-^RNH-NiOHi-R!-»
—> RN i N Ri + H2O
Радикалы R, R1( R2 могут принадлежать к жирному, ароматич. и гетероциклич. ряду; наибольшее значение имеют те соединения, в которых по крайней мере один из радикалов относится к ароматич. или гетероциклич. ряду. Обязательным условием для реакций (А) и (Б) является подвижность водородных атомов метильной группы, вызываемая специ-фич. свойствами соединения. Наиболее простые примеры конденсаций по типу (А), обусловленных подвижностью водородных атомов метильной группы, известны в ряду жирных соединений. Так, уксусный альдегид конденсируется с самим собой (I) в кротоновый альдегид, с ацетоном (II) в этилиденаце-тон и с нитрометаном (III) по схемам
I. СНз-СНО + СН3 СНО->СН3 СН(ОН) СН2·
•СНО -> СНз сн : СН СНО
II. СНз-СНО + СНз-СО-СНз -> СН3 СН(ОН)·
•СН2-СО-СНз -» СНз-СН : СН-СО-СН3
III. СН3 СНО + CH3N02-> СНз-СН(ОН)·
•CH2N02 -> СНз-СН : СНМ02
Во всех этих соединениях подвижность водородных атомов метильной группы вызывается соседством карбонила или нитрогруппы.
Эти соединения м. б. представлены и в энольной форме или в ациформе, так что подвижность водородных атомов в метильной группе объясняется возможным переходом водорода из метильной группы в соседнюю группу карбонила или в нитрогруппу по схемам
СНз-СО-СНз £СН» i С(ОН)-СНз
CHsN02 ii СН2 : Nf
хОН
Можно в силу этого предположить, что водо-роды свободной или замещенной метильной группы будут достаточно подвижны и в других соединениях, где есть возможность подобной кетоэнольной изомеризации соединений или изомеризации в ациформу, а также и других реакций, где допускается переход водородных атомов из метильной или метиленовой группы в другие группы. Такие соединения должен быть вообще способны к конденсации с карбонильными производными. Они м. б. выведены из простейших, вышеприведенных групп —СН2 · СО— и —СН2 · N02, если метильная (метиленовая) группа последних будет отделена от карбонильной (или нитрогруппы) сопрягающей цепью ви-ииленовых групп по схемам
,.0
—СН2 · (СН : СН)х СХ- и —СН2-(СН : CH)xN02
Подобную группировку мы встречаем в нитроах с нитрогруппами в о- и п-положеииях
70Η=0Η4
СЩ. ХС-СН3 и N02
^СН-С^
N0»
/СН-СН.
W
^сн-сн^
с—сн
3
к-рые в силу этого обладают достаточной подвижностью водородных атомов метильной группы, возрастающей с увеличением числа нитрогрупп. Так например, уже 2,4-динитро легко конденсируется с альдегидами по следующей схеме он
no2
/СН3
XNO«
О-СН Е -
N0
CHa-CH-R
/V
no2
NO
,CH : CH-R
no2
Подвижные водородные атомы имеются и в индоксиле и др. аналогичных соединениях, которые в кетоформе легко конденсируются с карбонилсодержащими соединениями, например с изатином (смотрите Индиго) или с глиоксалем, давая любопытный краситель
| ,/NH4 | /NH. | |
| СН2 4 | - осн-сно + сн» | |
| х с | х с | |
| II | II | |
| 0 | 0 | |
| ΝΗχ NH | ||
| -* ( | с=сн-сн=с | |
| к | А С | χ с А |
| II | II | |
| 0 | 0 | |
х>
Подвижность атомов водорода в данном случае обусловлена близостью СО-группы, так как последняя создает возможность кето-энолтаутомерии.
Подобная же таутомерия возможна и в /3-нафтоле и в любом другом продукте соответствующего строения. Но и α-нафтол может, сопрягаясь через виниленовую группу, по схеме он
I о
/СЧ 4 (СН
,^-сн
хсн
/Ч,
сн
Ч/СН
I
сн2
давать достаточную подвижность водородных атомов метиленовой группы в кетоформе, что позволяет проводить конденсацию с карбонилсодержащими соединениями по нижеследующей схеме (для простейшего индолигнона)
/-ч /-ч ч_ V
о—с ч сн2
сн=сн
| /— | —ч |
| ч_ | _ |
| ч | |
| -»о-с | с |
| ч |
сн=сн
0=С NH -
хс/
Ч-
-С NH ·
|ЧС
они о
/-
ч_
о=с ч /-
. ч__
ч 1 с=с,
Х<К сн=сн !
о
В ряду гетероциклич. соединений мы наблюдаем ту же подвижность водородных атомов метальной (метиленовой) группы, если по соседству с ней (или отделяясь через сопрягающую виниленовую группу) стоит гетероатом, преимущественно азот, способный отдавать свои валентности Еодороду метальной группы. Такую картину мы имеем в ме-
тилпиридине, а особенно в а- и у-метилхино-линах. Так, хинальдин и лепидин способны к изомеризации по схемам
-снч
Чс
/СН>н
N/C-CH*
СН3
4nh/c=ch2
С^сн. Ч^сн<
СН2I
СЧ
•N^— ^XNH и обладают достаточной подвижностью водородных атомов метильной группы для конденсаций с альдегидами (I) и (II). Аналогично ведет себя и мезо-метилакридин (III), который является фенопроизводным лепидина. Все эти продукты (кроме, естественно, β-ше-тилхинолина, неспособного к изомеризации) реагируют с альдегидами по следующим схемам (написаны упрощенно)
сн т/сн
0CH-R
Ч
СН3
IX. N.
/~
ч_
"Ч
_
- OCH-R -> N
ч_
N
~Ч
-
ч
NCH : СН · R
-СН : CH-R
III.
N<_
/“
Ч_
Ч_
ч
_ж
-СН : CH-R
Ч_>СИз
/—Ч
ч_
Ч—CH3-LOCH · R->N(
Ч
Ч Ч
Все они могут реагировать с любыми альдегидами. Особенно ценные П. п. получаются, если и радикал, связанный с альдегидом, имеет ароматич. или гетероциклич. характер. Так, П. п. для синтеза индиго (смотрите) по Байеру и Древсену получается при конденсации о-нитробензальдегида с ацетоном ,сно
+ СНаСО-СНа-»
xno2
,СН(ОН) СН2 СО-СНз /V /Сн : CH-CO-CH,
71·
no2 v nno2
Подвижность водородных атомов метильной (метиленовой) группы позволяет конденсировать продукты, содержащие эти группы, также с соединениями, содержащими нитрозогруппу—N0 по ур-ию(Б). Наиболее простой тип таких конденсаций в жирном ряду—это конденсация первичных нитропарафинов с азотистой к-той в нитроловые кислоты N02-CH2 + 0 : Ν·0Η -> Ν02·0Η·Ν(0Η)2 -»
ll R
-►Ν02·0 : N-OItSNOa-CH-N : О
it R
и вторичных нитропарафинов в псевдони-тролы по схеме
N02-CH + 0 : N-OH-s- N02—С—N-OH II I
(Ra) (R2> OH
N02—C—N : O -> 11
(Rs)
Более сложный пример—конденсация аце-тоуксусной кислоты (кетоэнолтаутомер) с азотистой к-той по схеме сн3—со
or—со
ХСН2 + 0 : N-OH -
СНз—СО OR—СО
NOH;
СНз—00 OR—СО СНз—СО. OR—СО
ХСН—N—ОН-
I
он
/CH-NO
Соответственно и другие кетоэнолтаутоме-ры, как например фенол, нафтолы и тому подобное., могут давать с азотистой кислотой продукты конденсации — нитрозопродукты по схеме (для /J-нафтола)
| 4 | · | ч- | ||
| ч | /, | |||
| 4 | ч | |||
| CH | CH2- | 0 : Γν | Ϊ-ΟΗ-* СН | СН—N—ОН-» |
| Чн- | -с
II |
Чн- | ~п 1
II он | |
| 0 | 0 | |||
| ч | ~ч | |||
| 4 | ч | |||
| ч | ч |
ч
->СН С—NOH СН С—NOH
Чн-Ч Чн-с
Ч
о
/---ч ч__
ч
СН С—N0
Чн-ОГ
I
он существующие в различных формах.
Соединения с подвижными водородными атомами метальной группы могут вступать в конденсацию и с другими соединениями, содержащими нитрозогруппу, в частности с ароматич. нитрозосоединениями, давая в результате конденсации азометины. Так например, 2,4-динитро, обладающий, по вышесказанному, подвижными водородами метальной группы, дает с и-нитрозодиметиланили-ном продукт конденсации—азометин
№2-<^>-Снз - О : N-<^>—N(CH3)3 -> >no2
NOa-<^>-CH2-N-<^>~N(CH3)2 ->
xno2 oh
-+ N02-<^>-CH=N-<(^^N(CH3)3
xno2
Аналогичные азометины м. б. получены и по ур-ию (В) конденсацией соответствующих альдегидов с аминами. Так, вышенаписан-ный азометин м. б. получен при конденсации 2, 4-динитробензальдегида с и-аминодиме-тиланилином
NO,-<^)-CHO +H2N-<^>-X(CH3)2I
no2
-» N02-O^CH(0H)-NH-<3^N(CH3)2
I
N02
-) N02-<^>-CH : N-<^>-N(CH3)2
I
N0„
Амины вообще вступают в подобные конденсации легче, чем метальные производные, т. к. водороды аминогруппы более подвижны и не требуют наличия в молекуле других групп, способствующих их подвижности. В силу этого конденсация аминов, особенно ароматических, с альдегидами идет весьма легко и часто применяется при производстве П. п. Продукты этой конденсации—а з о м е-т и н ы, или основания Шиффа, получение и строение которых м. б. выражено схемой Ar-NHj + ОСН R ^ Ar-N :СН R;
они отличаются небольшой стойкостью и расщепляются при действии к-т обратно на альдегид и амин. Эта нестойкость соедине ния способствует также и иным реакциям азометинов, в частности их восстановлению и присоединению элементов синильной кислоты (смотрите выше). Кроме того азометины способны к реакциям дальнейшего присоединения аминов и последующих перегруппировок в производные полифенилметанов.
Другой тип такой конденсации—конденсация гидразинов с кетонами по схеме
R · R
NHR-NH2 + 0:c Ч NHR-N : с 1 xR2 xR2
приводящая в случае дикетонов к пиразоло-вому циклу. Амины способны также вступать в аналогичные конденсации с соединениями, содержащими нитрозогруппу—N0; например при конденсации анилина с нитрозо-бензолом по ур-ию (Г) получается азобензол.
Образование азометина из анилина и бен-зальдегида
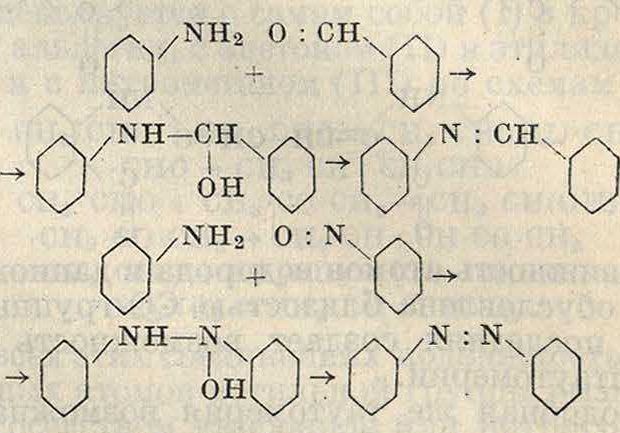
обнаруживает полную аналогию между этими процессами. Еще резче выступает аналогия между альдегидами и нитрозотелами,. если сопоставить с этиленовыми производными альдоксимы и диазогидраты
R-CH : СНОН, R CH : NOH, R-N : NOH, которые все способны к цис- и транс- (син- и анти-) изомерии. К данной группе реакций необходимо отнести также реакции, идущие по· схеме (Г), конденсации аминов с азотистой к-той ОН · N0, которые обыкновенно пишутся упрощенно
N
III
R · ΝΗϋ · НС1 + ОН · N : О -» R-N : NC1 или R—N—С1
(смотрите Диазосоединения), но с точки зрения вышеизложенных положений должны трактоваться более сложно. Любой амин (жирный или ароматич., даже неорганический) реагирует с азотистой к-той, образуя нестойкий продукт присоединения
| Ri | Ri | OH |
| /Н
R2-N< + 0 : 1 ХС1 |
R2x i
: N ОН >N-Cl7 1 |
1
-N 1 |
| R3 | R3 | OH |
Если в этом промежуточном соединении хоть один из радикалов представляет водородный атом, то возможны выделение элементов воды и образование более прочной двойной связи между атомами азота и дальнейшие реакции, обусловленные свойствами амина. Для аммиака реакция эта м. б. представлена следующей схемой н он
Н. i I -НС1 ХН3 + НС1 + NO-OH^ >N—N ----*.
CV i I
н он
Η он
- НС1 I I -2H20
-> N-N--*N“-N.
I I
H OH
Первичные алкиламины вследствие непрочности связи алкильного остатка с основным азотом дают нестойкие промежуточные соединения, распадающиеся с образованием а по схеме н он нч i i н
/N—N -* >n=n^-hci+n--:n + roh
СУ I I СУ I I Е ОН Е ОН
Первичные ариламины, в которых связь кислого арильного остатка (Аг) с основным азотом более прочна, дают более стойкие диазо-ниевые соли, которые лишь при нагревании переходят (с элиминированием азота) в фенолы по схеме
Н он
Нч I I н
—N -> >N=N СУ II СУ I I Аг ОН Аг ОН-
С1
N=N
I
Аг
НС1 + ΝΞΝ+Αγ-ΟΗ
R
Вторичные амины, жирные, жирно-ароматические и ароматические, дают при выделении воды соли нитрозаминов, легко гидролизующиеся в нитрозамины Rj он R! Rt
>Tsr—Ν’ -> Cl—A—N -» N—NO + HC1 СУ I I II I
r2 oh r2 oh r2
В третичных аминах невозможно выделение воды и они вообще не взаимодействуют с азотной к-той. Но если один из радикалов ароматический, то в нем весьма подвижны водородные атомы в «.-положении и двойная связь ядра активна, в результате чего выделяется вода и образуется (в перекисной форме) соль хиноноксима, переходящая далее в нитрозопродукт
R
С1 он
N—N
R
.Λ
О
он
С1 i I
>V
П-
С1
-Н
/Ч-ОН i H HO—N -
Rj
НО—N
или в хиноидной форме
Xl
N—Cl
N
НС1
Проведение этих реакций в технике см. Диазосоединения.
В. Реакции внутри молекулярной конденсации. Этот класс реакций получения П. п. отличается от предыдущих тем, что конденсация идет внутри молекулы самого соединения, в к-ром уже имеются все необходимые группы. В результате конденсации может образоваться либо новая прочная связь между группами или между одной из реакциеспособных групп и ядром соединения, приводящая к образованию или, как говорят, замыканию нового цикла, либо непрочная связь какой-либо группы с двумя различными положениями, приводящая в результате к образованию более стойкого соединения, содержащего эту группу уже в ином положении,—то есть к перегруппировке этой группы. Обычно реакциям замыкания цикла и перегруппировки, то есть внутримоле кулярной конденсации, предшествует одна из реакций предыдущих классов, в которых замыкающая цикл или перегруппировывающаяся группа вводится в соединение.
1) Реакция замыкания цикл а— циклизация—фактически сводится к введению группы в ядро соединения или в его боковую цепь, причем вводимая группа уже заранее присутствует в молекуле, связанная с одним из положений данного соединения. Легкость замыкания цикла зависит от пространственных натяжений и взаимного пространственного расположения замыкающихся частей молекулы. Легче всего замыкаются пятичленные и шестичленные циклы, высокая прочность которых подтверждена тео-ретич. расчетами А. Байера. При замыкании цикла можно предположить, аналогично предыдущему, промежуточную стадию присоединения внутри самой молекулы, обусловленную этими пространственными натяжениями и активностью двойной связи соединения. Из этой промежуточной стадии при действии водоотнимающих, кислотосвязующих или щелочесвязующих веществ выделяются элементы воды, кислоты или аммиака. Ниже рассмотрены наиболее употребительные реакции циклизации.
Замыкание цикла дигидроакридина при действии кислоты (иногда под давлением) на производные ди-о-диаминодифенилметана
^/VCHN л +hci2
^/χΝΗ2 ΝΗ2/ΧΧ ^ΧΝΗ/ίγ^
η2ν η η
/0Η2χ
ΝΗ
+ NHiCl
приводит к ценным Π. п., переходящим в акридиновые красители.
Аналогично ди - о - диоксидифенилметано-вые производные при нагревании с серной к-той, или иногда при нагревании до переходят в ценные П. п. для синтеза ксан-теновых красителей, замыкая цикл ксанте-на по схеме
/СН;
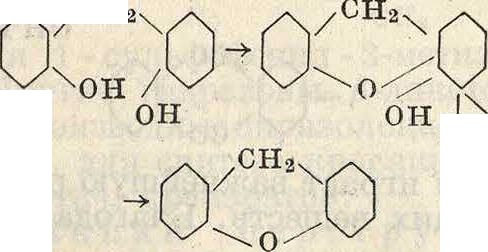
н н
Соответственные диамино- и диоксипроиз-водные дифенилкетона замыкают цикл акри-дона и ксантона по схемам
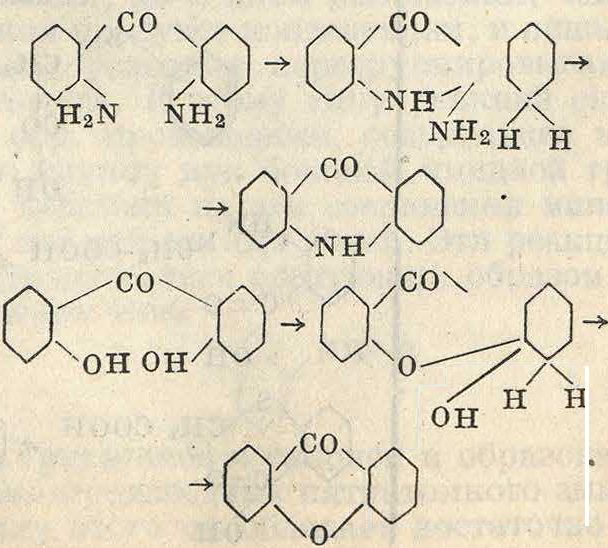
Точно так же ди-о-диаминопроизводные и диоксипроизводные дифенила замыкают циклы карбазола (дифениленимина I) и дифе-ниленоксида по схемам (I) и (II)
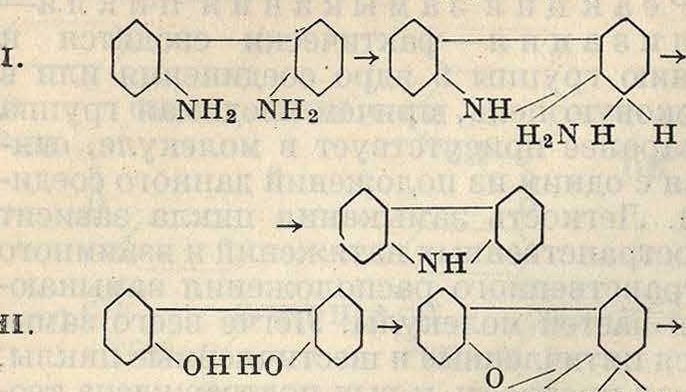
но он н можно получать не только синтетич. антра-хинон, но и ряд его производных. Вообще производные антрахинона м. б. получены одним из трех путей. По первому—заместители вводятся в готовый антрахинон, полученный окислением антрацена или синтетически из фталевого ангидрида и бензола (вышеприведенной конденсацией бензоилбен-зойной к-ты). По второму пути необходимые заместители уже находятся в тех производных, которые служат для получения производных бензоилбензойной кислоты; так например, 2-хлорантрахинон может быть получен конденсацией фталевого ангидрида с хлорбензолом в две стадии (промежуточный продукт—хлорбензоилбензойная кислота)
ссо
ч0
Весьма большое число реакций циклизации имеет место при наличии карбоксильной группы. Так например, акридон может быть получен при внутримолекулярной конденсации о-карбоновых кислот диариламинов (для простейшего случая дифениламинов) по следующей схеме
ос
I II-
СООН
/NH4^4
чГ н он н
СО
,ΝΗ.
хсо//
Эта реакция имеет широкое применение при получении смешанных акридонов, например ан-трахиноннафтакридона.
Соответственно о-карбоновая к-та дифе-нилкетона, т. н. бензоилбензойная к-та, замыкает при действии серной кислоты цикл антрахинона по схеме хо
#s Λ ^/соч00,
н он н
/соч Nx>
Эта реакция играет важнейшую роль в синтезе красящих веществ. Благодаря ей воз-
соон V
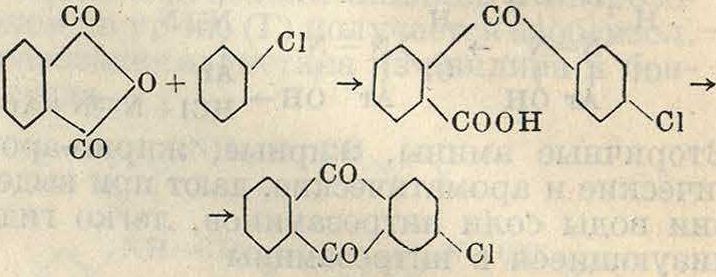
По третьему пути необходимые заместители вводятся лишь в заранее приготовленную бензоилбензойную к-ту и остаются в антра-хинонной молекуле после замыкания цикла, например
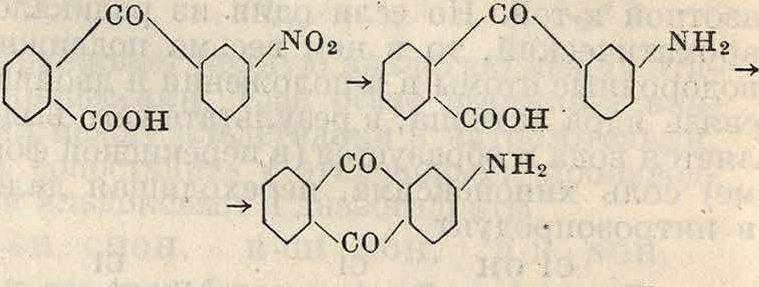
Различным сочетанием этих трех путей можно синтезировать целый ряд производных антрахинона, получение которых из готового антрахинона является затруднительным или невозможно.
Весьма большое значение имеют реакции замыкания циклов индола и тионафтена, приводящие к ценным промежуточным продуктам, применяемым для синтеза индиго и индигоидных красителей.
Ниже приведены примеры замыкания подобных циклов, происходящего при сплавлении с щелочами или иногда при действии кислых водоотнимающих средств и идущего соответственно двум типам
II.
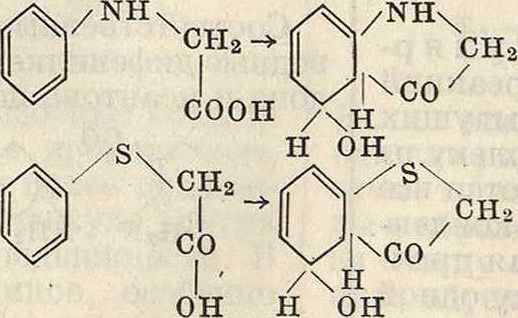
о
NH.
СО·7
-S
cm
СО
cm
,ΗΝ
сн2-соон
О
sc=o
ч он
/δχΟΗί,·
4 0=0 ч он соон
ΥΝΗ4
сн ·соон
/чс
/ч но он
/8ч
/NHX
снсоон сн ·соон
хс
/ч но он
хСо
/®ч снсоон
чСо
Ценные Π. п. получаются при конденсации аминов с глицерином или с акролеином по методу Скраупа. При этом под влиянием серной кислоты идет отнятие воды из алкильного производного амина, и получившиеся при этом производные дигидрохинолина окисляются введенным в реакцию окислителем. Общая схема реакции н он
,он сн2
ч
nh
сн-он
I
сн2
/СН*.
7 хснон Чтг I
Ч
NH
/СН2
/СН2
7 хснон
-
/СН2NH 7
ч юн.
XNH/
ЮН
I
сн2
/СНхХсн
N
*
I
сн
При замене глицерина или анилина в первой стадии реакции другими оксипроизвод-ными или аминами получаются различные производные хинолина. Аналогичная реакция идет и при конденсации антранола с глицерином, когда алкильное производное антранола замыкает при действии серной кислоты цикл бензола, образуя тем самым ценный П. п.—бензантрон—по схеме
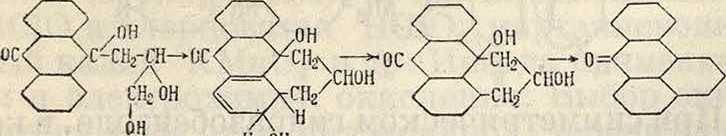
Ценные П. п. получаются из ацетиламино-антрахинонов, благодаря реакциеспособно-сти водородов метильной группы,стоящей по соседству с карбонилом, и кетогруппы антра-хинона. Так, в технике из ацетил-1-мономе-тиламиноантрахинона получают А-метил-1, 9-антрахинон-а-пиридон о
СН3
О
со I
N-СНз
οόό * ССО
и с
птт СН2 N-СНз
υ±1 ι i
С /А h2so4
о
II
с
/ч
СН N-CH,
!! I
/СЧ;
Хс о
Иногда при замыкании получаются циклы, содержащие более одного гетероатома. Так, при ацилировании о-диаминов, о-ами-нофенолов и о-аминотиофенолов получаются производные имидазолов, оксазолов и тиазо-лов по схемам /NH4
4 СО СНз
4NH2
,ΝΗ
,ЧИЧ /СНз
NH
чон
•ос
/NHX4
С-СН3
| ,/ΝΗχ Г | ΝΗ |
| с-сн -*[ | 1 |
| хон jj х | о -- |
С-СНз
I
он
/*Ч
с-сна
/NH
хс-сн3
* л
•о
/NH4
/N.n
С-СНз
4x8/он ч с-
хя
СНз
Изомерные циклы, содержащие два атома азота рядом—пиразоловые циклы,—получаются в результате внутримолекулярной конденсации в гидразонах /З-дикётонов по схеме о OR1 он
Ч I /OR1
R-NH С R—N—С<
I I n=c-ch2I
R2
R—N—С:
I
N=C
СН2 -
А=с
i,
R-
СН2
I
R2
+ R1OH
Так замыкается гидразон, полученный из и-сульфофенилгидразина и ацетоуксусного эфира, например о
Η II ЮС2Н5
ю- I сч
80зН-О СН2 ^
N=CX
СН3
ОН
i
N=C
I
СН;
ОС2Н5
СН2
• so3H
-о-г
3
=С7
ч
4СН2
сн3
образуя 1 - сульфофенил - З-метил-5-пиразо-лон. Другие гидразоны β-дикетонов дают также производные пиразолона—весьма ценные П. п. для синтеза красящих веществ и фармацевтич. препаратов.
2) Реакции перегруппировки представляют такую внутримолекулярную конденсацию, когда вступающие в нее группы находятся довольно стойко в том же соединении, но в ином положении, чем в конечном продукте конденсации, и лишь в спе-цифич. условиях перегруппировываются в последнее. К этому типу реакций склонны гл. обр. производные, содержащие какую-либо группу при боковой имидной группе, при действии на эти соединения минеральных к-т или кислых солей. Эти реакции могут трактоваться следующим образом: в соединении типа
NH—R
азот трехатомен и склонен к образованию с к-тами производных пятиатомного аммония; в силу этого он обладает достаточно силь-
ными остаточными сродствами, частично насыщенными остаточными сродствами ядра и радикала R. При действии кислоты остаточные сродства азота ослабевают, и связь его с R также ослабевает, в силу чего у R появля-. ются остаточные ненасыщенные сродства. С другой стороны, у фенильного ядра появляются те же сродства, передающиеся через конъюгирующие виниленовые группы к о-или «-углеродному атому соединения, которые насыщают сродства радикала R. В результате этого получается нестойкий цикл, к-рый в специфич. условиях разрывается, причем радикал R оказывается прочнее связанным с углеродом ядра по схеме (для «-положения)
н АС
Н—N—R Η—N—R Н—Ιί
I I !
н
| Н Ас | Н Ас
Ч |
| H—N— 1 | — NH NH· |
| 0 | ( и i
vOO |
| /-~
н |
/Ч 1 H R R |
Все до сих пор известные реакции перегруппировки такому толкованию реакции не противоречат. Так, фенилгидроксиламин переходит при действии серной кислоты в и-аминофенол
ΝΗΟΗ NH2
I I
I
он
Монометиланилин при нагревании под давлением с соляной к-той переходит в смесь о- и и-толуидинов
NHCH3 NHa NH2
1 /СНз
I
СНз
Нитрозамины вторичных жирно-ароматич. и ароматич. аминов переходят при стоянии с овым раствором хлористоводородной кислоты в и-нитрозопродукты
К—N-NO R—NH
о - ό
NO
Ациламины перегруппировываются при нагревании с ZnCl2 в кетоны NHCO—R NH2
J I
Vco-R
пере-
Соответственно сульфаминовые кислоты ходят в сульфокислоты аминов NH-S03H nh2I
О
S03H
Продукты присоединения амина к азоме-тину перегруппировываются в производные диаминодифенилметана
/ΝΗ-ΟΗϋ-ΝΗ.
/°Η2χ
NH v v4NH2
Диазоаминотела переходят в аминоазокра-сители
-NH-N: N-R « n:N-R
/г
nh2
Наибольшее технич. значение имеет эта реакция в применении к гидразотелам, которые подвергаются бензидинной и семидинной перегруппировке. Так,гидразосоединение типа
NH--NH
I
переходит в производное дифениламина, подвергаясь при действии НС1 тонна н. семидинной перегруппировке по схеме
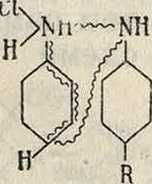
Нч
- NH.
а.
При симметрическом гидразобензоле, в котором оба «-положения свободны, мы имеем вполне равноценные части, в которых все остаточные сродства сконцентрированы у «-углеродных атомов, вследствие чего имеет место бензндинная перегруппировка по схеме
-NH H>Nci£!~N-H
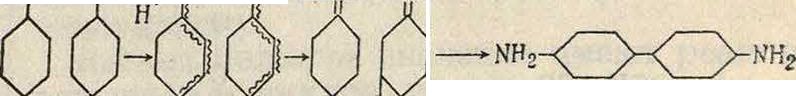
ΝΗϋ
-0-0-
νη2
NHi
НЧ—J ц
Аналогично получаются о-толидия и ди-анизидин ч ,сн3 сн3 осн3 осн3
ценные П. п. для синтеза субстантивных азокрасителей.
Нек-рые оксипроизводные также склонны к подобным перегруппировкам. Так, эфиры фенолов (угольный, уксуснокислый и прочие) при нагревании переходят в соответствующие кислоты или кетоны по схеме ,Ο-COOH Λ /ОН
/0-C0-R
COOH
,он
COR
Первой реакцией пользуются при получении карбоновых к-т фенолов (смотрите выше), причем реакция в присутствии NaOH приводит для простейшего фенола к салициловой к-те, а в присутствии КОН—к и-оксибензойной кислоте. Соответственно получаются и карбоновые кислоты нафтолов.
II. Реакции окисления и восстановления.
Эти реакции находят применение при получении самых разнообразных П. п. и сводятся при реакции окисления к обогащению молекулы соединения атомами кислорода или к уменьшению числа атомов водорода в нем, а при реакции восстановления—к уменьшению содержания кислорода или к обогащению соединения атомами водорода. Иногда имеют место одновременное окисление и восстановление соединения, когда его элементарный состав не изменяется, но одна из имеющихся в соединении групп окисляется за счет другой, которая при этом восстанавливается. Часто окисление и восстановление приводят к разрушению скелета соединения и к образованию менее сложных П. п.
1) Реакции окисления. Для проведения реакции окисления необходим окислитель, т.е.богатый кислородом реагент, отдающий свои атомы кислорода, которые либо внедряются в окисляемое вещество либо связывают водород последнего в виде воды. Наиболее употребительными окислителями являются: кислород воздуха 02 (чаще в присутствии катализаторов), озон 03, перекись водорода Н202, серный ангидрид S03, соли надсерной кислоты H2S20, (персульфаты), азотная кислота HN03, перекись марганца Мп02, перекись свинца РЮ2, хромовый ангидрид Сг03 и соли хромовых кислот (хро-маты и бихроматы), кислоты хлорноватистая НСЮ и хлорноватая НС103, марганцовокислый калий КМп04 и др. Иногда применяется и электрохимич. окисление. Выбор окислителя зависит от желательной степени окисления, с одной стороны, и от условий реакции—с другой; кроме того решающую роль играют и экономии, факторы. На ход реакции окисления кроме характера окислителя влияют также и ί° реакции и ее среда. Так, нек-рые реакции должны вестись в кислой среде, в то время как другие П. и. получаются при окислении в нейтральной или щелочной среде. В ароматическом соединении любая боковая углеродсодержащая цепь м. б. окислена, переходя через различные стадии окисления, в карбоксильную группу. Напр. при окислении а (I) в зависимости от условий реакции мы можем получить бен-зиловый (II), бензойный альдегид (III) и бензойную к-ту (IV) по схеме
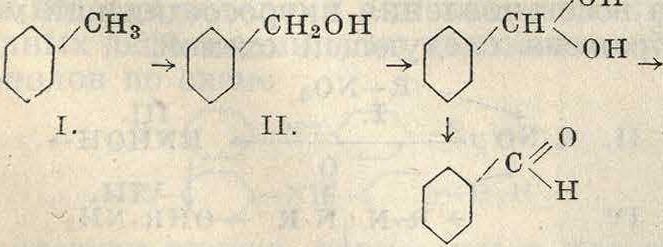
III.
/ОН
/CAlH Y VoH
R
R,
>СН2
N
к
/ОН R
снон -
Rj7 ЧОН Ri Производные трифенилметана м. б. окислены лишь в трифенилкарбинол по схеме с=о
R χrAch. r2
Rj-COH R2
В зависимости от выбранного окислителя, среды и ί° реакции можно вести реакцию так, чтобы получить максимальный выход желательной стадии окисления. Так, окисляя рассчитанным количеством перекиси марганца в сернокислой среде, получают удовлетворительные выходы бензальдегида, в то время как применение более энергичных окислителей или более жестких условий окисления приводит к бензойной к-те. Большого интереса заслуживает каталитич. окисление а кислородом воздуха на ванадиевом катализаторе, причем получается удовлетворительный выход бензальдегида.
Кроме окисления в боковой углеродсодержащей группе ароматич. и гетероциклич. соединения способны к окислению и в самом ядре. Так, можно ввести гидроксильную группу в ядро соединения прямым окислением по схеме
—СН ;":СН,
СН -> /С-ОН
-сн^· -СН7
Непосредственное получение фенола и нафтолов по этой схеме не нашло технич. применения вследствие недостаточности выходов; наиболее часто по этой схеме получают полиоксиантрахиноны по реакции Бонна и Шмидта (смотрите Бонна-Шмидта реакция) при действии высокопроцентного олеума на окси-антрахиноны в присутствии катализаторов, например селена.
Более жесткое окисление может привести к образованию хинонов и их производных. Так, антрахинон получается при непосредственном окислении антрацена хромовой кислотой
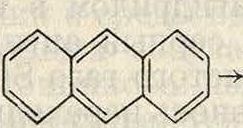
0
1
II
о
Аналогично получаются и фенантренхинон и аценафтенхинон, которые могут при более энергичном окислении переходить в соответствующие дикарбоновые к-ты—дифеновую и перинафталиндикарбоновую
0-0 ^ 0>-0 "·* О—О
_ _ I I
о=с с=о соон соон
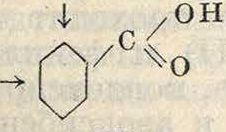
IV.
Точно так же дифенилметановые производные м. б. окислены в производные бензгид-рола и бензофенона по схеме н2с—О о < ( „ соон соон
Простейшие хиноны могут получаться при окислении соответствующих им диоксипро-изводных, но приготовляются чаще из более доступных аминооксипроизводных, проходя через стадию нестойких хинониминов по схемам
NH
О
NH2
I
OH
NH2
I
NH
О
II
№-бензохинон получается при окислении анилина в сильно кислой среде хромовой смесью по вероятной схеме
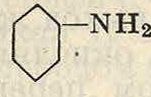
0-NHOH
чи он
|—nh2
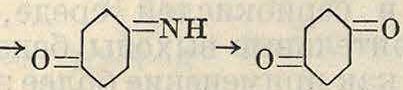
При совместном окислении и-аминоокси-и м-диаминопроизводных с аминами и фенолами в результате вторичной реакции присоединения к первичным хинонным продуктам получаются индамины и индофенолы (смотрите). Целый ряд красящих веществ класса метиленхинонных и хинонимидных получается в результате сложного окисления, протекающего через ряд фаз.
Весьма большое значение имеет окисление нафталина, сопровождающееся разрушением скелета нафталинового ядра и образованием, в зависимости от силы окислителя, фталоновой (I) или фталевой (II) к-т, причем последняя легко переходит во фталевый ангидрид (III)
00-0
| /COOH
г |
-ч,СООН | ч о о |
| хсо-соон х | хЧСООН ^ | -шо |
| I. | и. | III. |
Окисление КМп04 в щелочной среде приводит к образованию смеси продуктов, в то время как окисление хромовой к-той в кислой среде приводит исключительно к фталевой к-те. Значение последней в качестве П. п. для получения из нее антрахинонных красителей весьма велико. В конце 19 в технически осуществлено получение фтале-вого ангидрида при каталитич. окислении нафталина серным ангидридом в присутствии ртути. При этом серный ангидрид S03раскисляется до сернистого газа S02, к-рый в контактной системе вновь переходит в S03; иначе говоря, фталевый ангидрид получается из нафталина за счет кислорода воздуха. В настоящее время осуществлен метод прямого каталитич. окисления нафталина кислородом воздуха по методу Гиббса-Воля в присутствии ванадиевых катализаторов, состоящий в пропускании паров нафталина в смеси с воздухом через нагретые выше 400° трубки, содержащие катализатор. Успех в этом отношении позволяет синтетич. антра-хинону экономически конкурировать с ан-трахиноном из антрацена каменноугольной смолы. При окислении ароматич. соединений, содержащих в боковой цепи гетероатомы, действие окислителя обычно распространяется и на последние. Напр. при окислении анилина и других аминов получаются в первую очередь продукты окисления за счет аминогруппы (смотрите Черный анилин). Другие примеры окисления боковой гетероцепи даны в следующих схемах
RSH -> R-S-S-Rj -» RS03H R-S02H -> RS03H R-NH-NHa-s-RH rH20+JST2R-NH-NH-Rj -> R.N : N-R2
2) Реакция восстановления достигается обработкой соединения т. н. в о с-становителями. В качестве последних применяются различные химич. соединения, способные переходить в высшую степень окисления, а также нек-рые металлы, переходящие при этом в свои окислы или соли. Наиболее употребительны следующие восстановители: металлич. железо (в виде железных или чугунных опилок и стружек), цинк (гранулированный или в виде цинковой пыли), олово (в гранулированном виде); сернистые щелочи (Na2S, K2S) и сульфгидрат натрия (NaSH); соли сернистой кислоты (преимущественно в виде бисульфита NaHS03), соли сульфоксиловой и гидросернистой к-т в виде гидросульфитов (смотрите) и сульфоксила-тов различных марок; закисные соли железа; соли закиси олова (SnCl2) и треххлористый титан ТЮ13; весьма часто употребляется молекулярный водород (Н2) в присутствии катализаторов, например никеля или коллоидальных металлов. Значительно реже, преимущественно в научном исследовании, применяются металлич. натрий, амальгама натрия, фенилгидразин и др. восстановители, допускающие точную регулировку их действия. В зависимости от темп-ры восстановления и главным образом от условий среды имеется возможность регулировать степень восстановления, останавливаясь на необходимых стадиях.
В технике наибольшее значение имеет восстановление нитросоединений, нитрозосо-единений, хинонов и их производных (индамины, индофенолы), кубовых красящих веществ (в их лейкопроизводные), дисульфидных производных (в меркаптаны) и непредельных соединений в предельные (т. н. гидрирование). При восстановлении нитросоединений, подбирая условия среды (кислая, нейтральная или щелочная) и силу восстановителя, возможно останавливать процесс на желаемой стадии восстановления. Иногда промежуточные стадии, подвергаясь реакции конденсации, приводят к более сложным продуктам; иногда наблюдаются и реакции перегруппировки в промежуточной стадии, приводящие к новым продуктам. Общая картина восстановления нитросоединений м. б. изображена следующей схемой
_____—N02
I. III.
И. R-NO ---->· RNHOH-»
- о I
^--- и ; vili.
IV. -> R-N : N-R -+OHR-NHs
1
V. RN:N-R
I IX.
VI. R NH NH R -* NH2R R NH·.
I
VII. r-nh2
Так, нитробензол (I) при восстановлении в щелочной среде проходит через стадии ни-трозобензола (II) и фенилгидроксиламина (III). Последние, конденсируясь друг с другом, переходят в азоксибензол (IV), к-рый при дальнейшем восстановлении может перейти в азобензол (V) и гидразобензол (VI). Последний же, расщепляясь при нейтральном или слабо кислом восстановлении, пе-
реходит в анилин (VII). При восстановлении в слабо кислой среде или в нейтральной—железом в присутствии солей железа— реакция идет быстро до конца, до полного образования анилина. При восстановлении в сильно кислой (сернокислой) среде промежуточная стадия фенилгидроксиламина (III) не восстанавливается дальше, так как с большей скоростью претерпевает внутримолекулярную перегруппировку,переходя при этом в м-аминофенол
NHOH nh2
он
В технике щелочным восстановлением нитробензола и его производных пользуются для получения гидразосоединений, которые по выделении и обработке к-тами перегруппировываются в бензидин или другие производные диаминодифенила.
Нек-рые нитротела легко восстанавливаются сернистыми щелочами. Так, впервые анилин был получен Н. А. Зининым в Казани при действии сернистого аммония на нитробензол. Особенно часто пользуются этим методом, когда нужно восстановить лишь одну из нескольких имеющихся нитрогрупп. Так, из ж-динитробензола при действии сернистого натрия можно получить ж-нитроани-лин по схеме
NO240/NO2
Ароматич. амины и гидразосоединения можно получить и при восстановлении азосоединений, полученных методом азосочетания (смотрите Красящие вегцества). При этом цинк в щелочной среде приводит к образованию гидразосоединений, более же жесткие условия—к образованию двух молекул различных аминов по схеме
R-N: N-Rj-^R-NH NH R, -> R-NH2 + RrNH2Для получения ароматич. аминов служат также нитрозотела, переходящие при действии различных восстановителей в амины по схеме
R-NO R-NH-2
Для восстановления нитрозофенолов весьма удобными оказались сернистые щелочи. Те же сернистые щелочи оказались весьма удобными восстановителями для получения производных дифениламина из индаминов и индофенолов по схеме
r-0-n=o=Ri ’·
- R-<^>-NH-<^>~RjH
В последнем случае, равно как и при восстановлении азосоединений, мы имеем присоединение водорода к соединению, имеющему ненасыщенные связи. К этому же типу реакций следует отнести и образование вторичных аминов из т. н. азометинов (оснований Шиффа), представляющих аналоги азосоединений и получаемых при конденсации аминов с альдегидами по схемам н2
R-N : N-Rj-» R-NHNHR,
R-NHa + О : СН Ei -»
Н2
-> R-NH CHa-R,
Молекулярный водород в присутствии катализаторов также м. б. применен для целого ряда восстановлений, но его главное применение состоит в т. и. гидрировании (смотрите) непредельных соединений.
При восстановлении сернистой к-той или ее кислыми солями наряду с восстановлением часто имеет место и вступление сульфо-группы в ядро соединения. Так, нитрозо-/J-нафтол переходит при этом в 4-сульфокис-лоту 1-амино-2-нафтола
NHOH NHS
S03H
индофенолы переходят в сульфокислоты производных дифениламина с невыясненным до сих пор положением еульфогруппы.
Условия восстановления в бензольном, нафталиновом и антраценовом ряду весьма сходны. Лишь восстановление нитрогрупп в производных антрахинона вследствие большой их реакциеспособности идет значительно легче и протекает особенно гладко при нагревании со слабыми щелочными восстановителями, как станниты или сернистый натрий. Употребление более сильных восстановителей нежелательно, т. к. при этом происходит восстановление карбонильных групп антрахинона. Сам антрахинон легко переходит в оксантрол при действии Zn-пыли в щелочной среде.
Тиофенолы получаются часто из дитиопро-изводных восстановлением последних в щелочной среде цинковой пылью или железными стружками. Так, ценный П. и. для ряда тиоиндигоидных красителей—тиофенол-о-карбоновая к-та—получается при восстановлении т. н. дитиосалициловой кислоты /S s ^ 2 0/SH
ХСООН соон/^ V^COOH О”восстановлении кубовых красящих веществ см. Крашение. О технике восстановления нитробензола железом см. Анилин. Восстановление цинковой пылью в зависимости от свойств исходного и конечного продукта, от t° реакции и среды ведется либо в открытых деревянных сосудах либо в закрытых железных (оцинкованных). Нелетучие вещества восстанавливаются в большинстве случаев в открытых деревянных чанах, снабженных мощными мешалками. Иногда происходит одновременно с протекающей реакцией и окисление и восстановление вещества, причем одни группы вещества окисляются за счет других групп того же вещества, которые при этомвосстанавливаются; например о-нитро и его производные при нагревании их со щелочами переходят в антраниловую к-ту или ее ппоизводные по схеме
В этой реакции нитрогруппа окисляет метил до карбоксила, сама переходя при этом в > аминогруппу. Аналогичное внутримолекулярное окисление и восстановление мы имеем при получении нафтазарина из динитроан-трахинона (смотрите Красящие вещества синтетические).

-> R-N : ΘΗ-Ri
| Название | Формула | Получение | Свойства | Главное применение | |
| Нитробензол | 0 | —no2 | Из бензола нитрованием | Жидкость 1°пл.Λ з0 У кип“ 203° Dl6 1,208 | Для производства анилина, бензиди-на и как суррогат - миндального масла |
| Динитробензол
! |
ΝΟ,-ι | y* | Из бензола или нитробензола нитрованием | Иглы 1°пл- 90° t°Kun· 297° D 98 1,369 | Для производства ле-нитроанилина и jvt-фенилендиамина |
| о-Нитро | сн
1 0- |
3
-no2 |
Из а нитрованием вместе с п-изомером | Жидкость 1°кип· 220,4° В15 1,1643 | Для производства о-толуидина, толи-дина и нитробенз-альдегида |
| п-Нитроюлуол | СН3-( | Из а нитрованием вместе с о-изомером | 1°пл. 54,5°
V3кип“ 237,7° D 5 5 1,1392 |
Для производства η -толуидппа и стильбеновых красителей | |
| 1, 2, 4-Динитрото-
луол |
ΝΟϋ-Г
сн,—( |
y* | Из а нитрованием | t‘n“. 71° -^70,5 1,3108 | Для толуилендиа-мина, сернистых красителей и тротила |
| 1, 2, 4, 6-Тринитро (тротил) | ΝΟ,-Υ
сн,-( |
3№
1 «h |
Так же | 1°пл. 80,8° ает | ч. вещество |
| 1-Нитронафталин
(а-Н.) |
с | no2
ό |
Из нафталина нитрованием | Желт, кристаллы 1°пм 61° кип- 304° 1,331 | Для а-нафтнламина |
| 1 - Н птр оантрах иное (а-Н.) | а | ) no2
ίύ D |
Из антрахинона нитрованием | Желт, кристаллы Гпл. 232,5 233,5° кип“ (7 миллиметров) 270° | Для а-аминоантра-хинона |
| 1,5-Динитроантра-хинон | О no2
ii ! ссо Т 1! NOs, 0 |
Так же | Желт, кристаллы t%M>3S0· | Для 1, 5-диам-ноан-трах нона и других красящих веществ | |
| о-Нитроанилин | nh2
о- |
140-2 | Из оксанилида нитрованием и -1 и а минированием о-нитро-хлорбензола | Оранж, крист.
Г,.,. 71,5« Летуч е вод. парами |
Для красящих веществ и о-фенилен-диамина |
| Jt-Нптроанилин | №t | !
j— NO, |
Частичным восстановлением динитробензо.а | Желт, кристаллы t%.“. П4°
1 °кип- 285° |
Для азокрасителей |
| ?1-Нитроанилин | nh2 | j—NO,
i |
Из ациланили-лдов нитрованием или ами-нированием п-иптрохлор-бензола | Желт, кристаллы 1°пл. 147° | Для азокрасителей (холодное крашение) |
| Наввание | Формула | Получение | Свойства | Главное применение |
| 1, 2, 4-Динитроанилин | N02 -Α-Ν02NH2-IJ | Аминированием динитрохлор-
бепзола |
Желт, кристаллы (V“. 176» | Для сернистых красителей |
| о-Нитрофеноп | ОН
1 0-no2 |
Нитрованием фенола или гидроксили-рованием о-нитрохлор-бензола | Желт, кристаллы 1°пл. 45° о1°κιιη· 214°
D14 1,484 |
Для дианизидина |
| п-Нитрофенол
1 |
oh-0"° | Нитрованием фенола или гидроксилирова-нием n-нитр οχ лор бензооАа | Бесцветные кристаллы | Для п-аминофенола |
| 1, 2, 4-Динитрофе-нол | no2—A-no2
oh“U |
Омылением ди-питр ох лор бензола | Желт, кристаллы i°пл. П4° | Для производства сернистого черного |
| 1, 2, 4, 6-Тринитро-фенол (пикриновая к-та) | NOi—A— no2oh-U
1 NO 2 |
Нитрованием динитрофенола | Желт, кристаллы 1°пл· 122,5° | ч. вещество |
| о-Нитрохлорбепзол | Ci
Cr” |
Нитрованием хлорбензола | Желт, кристаллы 1°пл· 32,5°
243° 1,338 |
Для о-нитрофенола и его производных |
| п-Нитрохлорбензол | Так же | Кристаллы 1°пл. 83° t°Kim· 242° D22 1>380 | Для n-нитр офенола и я-нитроанилина | |
| 1, 2, 4-Дипитрохлор-бензол | №2 H02Cl/^ | Тан же | Желт, кристаллы 1°пл. 50° (48е) V*кип. 315° 1,Н07 | Для динитрофепола |
| Хлорбензол | (Г | Хлорированием бензола | Бесцвет. жидк. 1°пл. 45° 1°кип. 132 £>15 1,1125 | Для нитрохлорбен-вола
. |
|
Хлористый бензил |
0-CH2Cl | Хлорированием а без катализатора при освещении | Бесцвет. жидк. Слезоточивое средство 17 °
£>15 1,040 |
Для бензилнрова-ния и получения бензальдегида |
| 1-Хлорантрахиноп
(а-х.) |
0 Cl
II i ego ό |
Из сульфокислоты антрахи-нона | Желт, кристаллы 1°пл. 102° | Для антрахиион-ных красителей |
| 2-Хлорантрахинон
(Р-Х.) |
OCO“C
.0 |
Так же | Желт, кристаллы £ ° пл. 210° | То же |
Т. Э. m. XVIII.
8
| Название | Формула | Получение | Свойства | Главное применение |
| 1, 5-Дихлорантра-хинон | О С1
Сф С1 О |
Из сульфокислоты антрахи-нона | Желт, кристалль 1°пл. 248-261° | Для антрахинон-ных красителей |
| 2, 6-Дихлорантра-хиион | с.-С00-О!
II О |
Так же | Желт, кристаллы 1°пл· 292° | То же |
| Анилин | Q—νη2 | Восстановлением нитробензола | Жидкость
1°кип. 1°4° D15 1,0268 |
Для получения красящих веществ |
| о-Тодуидин | СНз
Q-nh2 |
Восстановлением о-нитрото-луола | Жидкость 1°кип. 199,4° D15 1,0037 Dго 0,999 | Для красящих веществ |
| п-Толуидин | A-nh2
СНз-U |
Восстановлением п-нитрото-луопа | Бесцв: крист. 1°пл. 42,8° 1°кип· 200° | Для фенилирова-ния красящих веществ |
| 1, 3, 4-КсиЛИДИН (ж-К.) | сн3
Λ-νη2 |
Восстановлением 4 -нитро-ж-ксилола | Жидкость 212° Dis 0,9184 | Для азокрасителей |
| 1-Нафтиламип («-н.) | nh2
03 |
Восстановлением а - нитронафталина | Кристаллы
t-V*. t°Kim. 300,8° Д20 1,171 |
Для азокрасителей и для сульфокислот |
| 2-Нафтиламин
(0-Н.) |
οα*ΝΗ· | Аминированием
0-нафтола |
Кристаллы 1°пл П2° оί кип· 291 | Для азокрасителей и для сульфокислот |
| 1-Аминоантрахинон
(а-А.) г |
0 nh2
φό 0 |
Восстановлением а-нитро-антрахинона или аминированием α-сульфо кислоты | Красн. кристаллы; 1°пл. 252—253° | Для антрахинон-ных красителей |
| 2-Аминоантр ахинон (β-Α.) | α!ο-“
II О |
Аминированием /3-су л ь ф ок ис л от ы антрахинона | Красн. кристаллы; 1°пл. 302° | Для индантрена н др. антрахинонных красителей |
| 1, 5-Диаминоантра-хинон | 0 νη2
φό ΝΗ2 0 |
Восстановлением динитро-антрахинона или аминированием дисульфокислоты | Красные иглы 1°пл .>320° | Для антримидных красителей |
| Название | Формула | Получение | Свойства | Главное применение | |
| Ацетанилид | Q—ΝΗ-СОСНз | Ацетилированием анилина | Бесцв. крист. 1°пл· | В медицине (антифебрин) п для получения нитроанилинов | |
| м-Фенилендиамин | nh2- | 0-nh2 | Восстановлением динитробензола | ^°пл. 63° t°icun- 237 Легко окисляется | Для красящих веществ |
| п-Фенилендиамин | nh2— | ^j-nh2 | Восстановлением п - нитроанилина или п-аминоазобен-зола | *°пл- 147° кип. 267° t°mn. (13 миллиметров) 150° | Для окраски мехов (уреол) |
| 1, 2, 4-То л у илсег диамин (jvt-T.) | nh2— | CH3
Гн- |
Восстановлением динитроа | t%u- 99° t кип. 280° Окисляется | Для ряда красящих веществ |
| п-Аминодифенил-
амин |
nh2-(^ | /NH.
г 40 |
Восстановлением нитродифениламина или азокрасителей | Гш- 66° (78°)
ί кип. 354 |
Для незелепо-черно-го анилина |
| Метилапилин | Q-NH-СНз | Алкилированием анилина и другими путями | Жидкость ^ кип. ^ 193,5° Б1б 0,9921 | Для красящих веществ и дляч. вещества (тетрила) | |
| Этиланилин | Q—NH-C1Hs | Так же | ЖИДКОСТЬ 204° Б1б 0,9648 | То же | |
| Диметиланилин | 0-N(CH3)2 | Алкилированием анилина | Жидкость ί теми.+193
i°3acw·+95° jDi6 0,9621 |
Для красящих веществ и для ч. вещества (тетрила) | |
| Диэтиланилин | 0-N(C2il5)2 | Так же | Жидкость t mm· 216 217° Dis 0,939 | То же | |
| Этялбензиланилнн | n-N<C-
4CH2.C6H5 |
Бензилирова-нием моноэтил-анилина | Жидкость V*кип. 285 286° | Для кислотных ме-тиленхинонных красителей | |
| Дифениламин | (Tt) | Из анилина и солянокислого анилина в автоклаве | Бесцв. крист. 1°пл. 64° | Для красящих веществ и в военном деле | |
| Фенилглицин | —NH—CH2
CO OH |
Конденсацией анилина с хлор-уксусной к-той или с формальдегидом и KCN | Кристаллы Гпл. 126—127° | Для синтеза инди-гоидов | |
| n-Амино фенол | OH— | rH- | Восстановлением п-нитрозо-фепола или п-нитрофенола | 1°пл. 186° | Для лекарств (фенацетин^ в фотографии и для красящих веществ
_а__i |
*8
| Название | Формула | Получение | Свойства | Главное применение ; |
| „и-Аминофенол | ОЯ-0—νη2 | Гидр окси лир о-ванием лс-суль-фокислоты анилина или другими путями | Бесцв. крист. 1°пл. 123° | Для красящих веществ |
| м-Диметиламино-
фенол |
oh-Q-n(ch3)2 | Гидроксилиро-ванием лс-суль-фо· ислоты ди-метиланилина | 1°пл· 87° t°Kun. 265—268° | Для родаминов |
| м-Диэтиламино-
фенол |
OH-0-N(C2H6)2 | Гидроксилиро-ваниехм л<-суль-фокисюты ди-этилапилина | 1°пл. 78°
1°кип. 275—280° |
То ше |
| Фенол | (Г | Щелочным плавлением бензол-сульфокислоты | Кристаллы Ь°пл. 42,5—43° пип. 181,3° | В медицине и для красящих веществ |
| Резорцин | ОН—0—он | Из ле-дисульфо-кислоты бензола | Кристаллы 1°пл· 119°
^ кип. 276,5° |
Для фталеиновых нитрозо- и азокрасителей |
| Пирогаллол | он
1 ОН-0—он |
Из галловой кислоты | 1°пл. 182° | В фотографии и для галеина |
| 1-Нафтол (а-Н.) | он со | Из а-нафтилами-на или из а-сульфокислоты нафталина | Кристаллы 1°пл- 94° t°кип. 278—280° | Для сульфокислот |
| 2-Нафтол (β-Η.) | аг | Из ^-сульфокислоты нафталина | Кристаллы Ь°пл. 122° ί°кип. 285—286° | Для азокрасителей и для сульфокислот |
| 1, 2-Диоксиантра-хинон (ализарин) | 0 он
С00“°" О |
Окислением и щелочным плавлением 0-сульфокислоты ан-трахинона | Желт, кристаллы 1°пл. 290° | 1
Как краситель и П. п. для антрахи-нонных красителей |
| 1, 4-Диоксиантра-хияон (хинизарин) | О он
. 0» о он |
Конденсацией фталевого ангидрида с гидрохиноном или п-хлорфенолом | Оранш. иглы 1°пл. 194° | Для антрахинон-ных красителей |
| 1, 5-Диокеиантра-хинон (антраруфин) | о он
Χό |
Щелочным плавлением 1, 5-дисульфокислоты антрахинона | Желтые блестки Гпл. 280° | То ше. |
| Ώ | - | i |
| Название | Формула | Получение | Свойства | Главное применение |
| 1, 3, 5, 7-Тетраокси-аптрахинон (антрахризон) | О ОН
0НОЮ-» оно |
Конденсацией двух молекул ре-зорцивкарбоно-вой кислоты | Оранж, иглы | Для антрахипон-ных красителей |
| 1
Салициловая кислота |
Ггон
1^—со он |
Из фенола и углекислоты | t°nA. 156,8° | В медицине и для азокраеителей |
| Аытрани лова я кислота | (Λ-νη2
соон |
Из фталевого имида | Кристаллы Х°пл. 145° | Для индигоидных красителей
• |
| о-Тиофено лкарбоно-вая кислота | p-SH
соон |
Из антранхтло-вой кислоты через диазосоединение | Кристаллы 1°пл. 163-164° Легко окисляется | Для тиоиндигоидов |
| Галловая кислота | он
1 он—Q—он 1 соон |
Гидролизом тан-нина | Кристаллы с 1 ч.
Н20. бесцв. Плавится с разложением при 222—210° |
Для галловых красителей и пирогаллола |
| 2, З-Нафтолкарбо-новая кислота | (Тгон
ls/kJ-cooH |
Из β - нафтола и С02 | 1°пл. 216° | Для нафтолов AS |
| п-Нитрозофенол | й
0 1 о 1 о W |
Нитрозирова-нием фенола | Раз л. при плавлении выше 125° | Для нитрофенолов и сернистых красителей |
| 1, 2-Нитрозонафтол | N0
агн |
Нитрозирова-нием 0-нафтола | Желтые кристаллы; t°nA. 110° | Нитрозо-краситель для аминонафтолов |
| п-Нитрозодиметил-
анилип |
N(CH3)2
NO-IJ |
Нитрозирова-нием ди метил-анилина | Соли желтые, основание зеленое; t°nA. 85° | Для красящих веществ |
| Бепзолсульфокис-
лота |
0-so3H | Сульфированием бензола | Кристаллы с 1 ч. Н20; Х°пл. без-водпой соли 53° | Для получения фенола |
| 1, З-Бензолдисуль-фокислота | S03H—0—S03H | То же | Кристаллы е 21/2 ч. Н20 | Для получения резорцина |
| 1-Нафталинсульфо-кислота (а-Н.) | so3H
06 |
Сульфированием нафталина на ХОЛОДУ | Кристаллы с 2ч. Н20; 1°пл. 90° | Для а - нафтола и дальнейшего сульфирования |
| Название | Формула | Получение | Свойства | Главное применение |
| 2-Нафталинстльфо-кислота (|8-Н.) | ог°-н | Сульфированием нафталина при 165° | Кристаллы с з ч. Н20; ί°τчл· 83° | Для ^-нафтола |
| 2, 7-Нафта линди-сульфокислота | so3h~QQ-so3h | Сульфированием нафталина дымящей серной кислотой | Растворимые соли | Для диоксинафта-лина и для дальнейшего сульфирования |
| 1,3, 6-Нафталинтри-сульфокислота | S03H
m S03H-lN/14/l— S03H |
Сульфированием нафталина крепким олеумом | Растворимые соли | Для Н-кислоты |
| 1-Антрахинонсуль-фокислота (а-А.) | 0 S03H
ΟΪ) n 0 |
Неполным сульфированием ан-трахинона в присутствии Hg | Сульфохлорид— 1°пл. 218°. Калиевая соль трудно растворима на холоду | Для антрахинон-ных красящих веществ |
| 2-Антрахипонеуль-фокислота (β-Α.) | C00~SO,B
1! 0 |
Неполным сульфированием ан-тфахинона | Сульфохлорид— 193°. Натр, соль плохо растворима | То же |
| 1, 5-Антрахинон-дису ль фокислота | 0 S03H
C00 [ II H03S 0 |
Исчерпывающим сульфированием антрахинона в присутствии Hg | Легко растворимые соли | Для антрахинон-ных красящих веществ |
| 1, 8-Антрахинон-дисульфокислота | H03S 0 S03H
i Ii i Сф 0 |
Так же | Соли труднее растворимы, чем 1, 5-А. | То же |
| 2, 7-Антрахинон-дисульфокислота | 0
II №000-8ОзН О |
Исчерпывающим сульфированием антрахинона | Легко растворимые соли | То же |
| 2, 6-Антрахинон-дисульфокислота | So,k-oJo-so·"
О |
Так же | Труднее растворимые соли, чем в предыдущем | То же |
| 1, з-Анилинсульфо-кислота (метанило-вая кислота) | nh2-Q-so3h | Восстановлением нитросульфокислоты бензола | Кристаллы С 11/2 ч. Н»0 | Для азокрасителей и для .и-аминофе-нола |
| Название | Формула | Получение | Свойства | Главное применение |
| 1, 4-Анилинсульфо-кислота (сульфани-ловая кислота) | nh2-
LJ-so3h |
Сульфированием анилина бакпро-дессом | Кристаллы с 2 ч. Н20 | Для азокрасштелей |
| 1, 4-Нафтиламин-сульфокислота (нафтионовая) | nh2
00 |
Сульфированием а -нафтиламина запеканием | Нерастворима в воде; соли растворимы | Для азокрасителей, для нафтолсульфо-КИСЛОТЫ (1,4) |
| 1
S08H |
||||
| 1, 5-Нафтил амин-сульфокислота | NHj
00 |
Восстановлением 1, 5-нитросульфокислоты | Плохо растворима в воде; соли растворимы | Для азокрасителей
. |
| so8h | ||||
| 1, 8-Нафтиламин-сульфокислота (пери-кислота) | H03s nh21 1
со |
Восстановлением 1, 8-нитросульфокислоты | То же | Для азокрасителей, для сернистых красителей |
| 2, 5-Нафтиламин-сульфокислоха (у-киелота Даля) | ОТ" | Сульфированием β-нафтиламина вместе с изомерами | Растворимость натриевой соли в е велика | Для азокраеителей |
| S03H | ||||
| 2, в-Нафтиламин-сульфокиелота (к-та Бреннера) | ΛΑ -nh2
S03H-IJJ |
Сульфированием β-нафтиламина вместе с изомерами | Плохо растворима; трудно растворима Na-соль | Для азокраеителей |
| 2, 7-Нафтиламин-сульфокислота | so,H-QQ-NHi | Так же | То же | То же |
| 2, 8-Нафтилампн-сульфокислота (баденская к-та) | so3H
ЙГ·. |
Так же | Плохо растворима в воде. Натриевая соль плохо растворима в у | То же |
| 1. 3, 6-Нафтил амин-дисульфокислота (к-та Фрейнда) | nh2
00 |
Восстановлением 1, з, 6-нитродисульфокислоты | Растворима в воде; кислая К-соль плохо растворима | То же |
| SOJH-/4v/4 4SOsH | ||||
| 1, 3, 6, 8-Нафтял-аминтрисул ь фоки-слота (к-та Коха) | H03S NHs
! Ί 00 " |
Восстановлением 1, 3, 6, 8-китротрисульфо-кислоты | Высаливается нацело NaCl в виде кислой динатр. соли | Для Н-кислоты |
| SOsH/4/S//4S08H | ||||
| 2, 5, 7-Нафтиламин-диеульфокислота | so3h-0Q-nh2 | Сульфированием β - нафтиламина вместе с изомером | — | Для азокраеителей и для 1-КИСЛОТЫ |
| w
0 |
| Название | Формула | Получение | Свойства | Главное применение |
| 2, 6, 8-Нафтиламин-дисульфокислота (амидо-(х-кислота) | S03H | Сульфированием | _ | Для азокраеителей, |
| 1
/γ -ΝΗ2 SOaH-lJJ |
/S-нафтиламина вместе с из омер ом | для у-кислоты | ||
| 1, 4-Нафтолсульфо- | он | Гидроксилиро- | Растворима в | Для азокрасителей |
| кислота (кислота Невиль-Винтера) | со
! S03H |
ванием нафтио-новой к-ты | воде | |
| 2, 6-Нафтолсульфо-кислота (кислота Шеффера) | Λ л /ОН
„.со |
Сульфированием /3-нафтола вместе с изомером | 1°пл- 125°- ДВУ-патриевая соль нерастворима в у | То же |
| 2, 8-Нафтолеульфо-кислота (кроцеино- | S03H
1 |
Так же | Двунатриевая соль растворима | То же |
| вая кислота) | or | в у | ||
| 2, 3, 6-Нафто л дисульфокислота | m-°H | Так же | Растворима труднее G-к-ты, | То же |
| (R-киелота) | S03H— 0 0 —S03H | легко сочетается с диазониямя | * | |
| 2, 6, 8-Нафтолдп-сульфокислота (G-кислота) | S03H
зо,н-0ГН |
Так же | Легко растворима; трудно сочетается с диазо-ниями | То же |
| 2, 3, 6, 8-Нафто л-трисульфокислота | S03H | Исчерпывающим | То же | Для 2, 3, в, 8-ами- |
| ίΥ|-°Η
S03H— —S03H |
сульфированием
^-нафтола |
нокислоты | ||
| 1, 2, 4-Аминонаф-толсульфокислота | SHj | Восстановле- | Дает стойкое | Для азокрасителей |
| (XT
1 S03H |
нием бисульфатом нптр.озо-/)-нафтола | диазопроизвод-ное, нерастворима в воде | - | |
| 2, 5, 7-Аминонафтол-сульфокислота | SOaH-ZV^-NHa | Щелочным пла- | Плохо раство- | То же |
| влением дисуль- | рима в холодной | |||
| (1-кислота) | UJ
1 OH |
фокислоты 0-на-фтиламина (2,
5, 7) |
воде, нерастворима в е | I |
| 2, 8, 6-Аминонаф-юлсульфо кислота (-/-кислота) | OH | Щелочным пла- | Нерастворима в | То же |
| 1
ΛΛ-ΝΗ, SOaH-CXJ |
влением дисульфокислоты /3-нафтил амин а (2, 6, 8)
* |
горячей воде | ||
| 1, 8, 8, 6-Аминонаф-толдисульфокислота (Н-кислота) | OH nh2 | Щелочным пла- | Слабо раствори- | То же |
| 1 1 00
S03H/X/X/XS03H |
влением кислоты Коха | ма в холодной воде |
| Название | Формула | Получение | Свойства | Главное применение |
| 1, 8, 2, 4-Амивонаф-толдиеульфокислота (Чикого SS-кис-лота)
. |
он nh2
аг,н ! SO,Η |
Щелочным плавлением три-сульфокислоты а-г,афтиламина (1, 2, 4, 8) | Хорошо растворима в воде
. |
Для азокрасителей |
| 1, 8, 4,6-Амияонаф-толдисулъфокисло-та ^К-кислота) | он νη2
I 1 soaH-00 1 S03H |
Щелочным плавлением три-еульфокислоты 0-нафтиламина (1, 4, 6, 8) | Суррогат Н-кис-лоты | То ше |
| 1,3, 5-Фенил мети л-пиразолон | 0
II CH2 —cx ! N~<Z>. C=N/ CH3 |
Копденсацией адетоуксусного эфира с фенил-гидразоном | 1°пл- 127° | Для лекарств и для пиразолоновых красителей |
| 1, 3, 5-Парасульфо-фенилметилпиразо-лон
ν’:ι |
0
II ch2-cn N—( ^>-SO,H Cr__:N 1 CH3 |
Конденсацией ацетоуксусного эфира с фенил-гидразинсуль-фокислотой | Для пиразолоновых красителей | |
| Индоксил (3-окс-индол) | Λ/ΝΗ
CH a OH |
Щелочным плавлением фенил-глицина | Кристаллы, легко окисл.
* ПА- 85 |
Для индиго |
| Окситионафтен
(Тиоиндоксил) |
/N/S
1 J CH AC<? 1 OH |
Щелочным плавлением фенил-тиогликольор-токарбоновой кислоты | 1°пл. 71° | Для тиоиндигоидов |
| Хинальдин
(2-метилхиполин) |
Λ /СНчЧ
П |H Vn„ ^с-°Нз |
Конденсацией анилина с ацетальдегидом | Жидкость кип· 246 248° D1° 1,059 | Для хинолиновых красителей |
| Фталевый ангидрид
;. -. |
n/CO»
xco/ |
Каталитическим окислением нафталина | Кристаллы 1°пл· 128° 1°кип- 284,5° | Для индиго и ант-рахинонных красителей |
| Фталевый имид | ^4/C0
NH ‘vAco |
Действием NH3на фталевый ангидрид | Кристаллы <%м. 233,5° | Для антраниловой кислоты |
| 2-Метилантрахинон
(β-Μ.) |
0.8-0
1 о к |
Конденсацией фталевого ангидрида с ом | i °пл. 177° | Для кубовых ант-рахинонных красителей |
| Название | Формула | Получение | Свойства | Главное применение |
| Бензидин | ОП
NH2/ks/l ^xNH2 |
Перегруппировкой гидразобен-
зола |
1°плЛ%8°· Сульфат плохо растворим | Для субстантивных азокрасителей |
| Толидип | снзч. _. /ОНз
NH/U UxNHa |
Перегруппировкой гидразото-
луола |
t°«A. 12 9° | То же |
| Диаыизидин | 0Η3° ^_κ /00Η8 νη/^ ^νη, | Перегруппировкой гидразоани-зола | 1°пл. 13 | То же |
| о-Нитробензаль-
дегид |
О7-0·
V CH 11 0 |
Из нитробен-зальхлориДа или другими путями | 0 1°пл. 43,5° оt° кип Λ 23 миллиметров) 153° | Для индиго |
| Оксибензальдегид | сно
Ο-он |
Из л«-амино-бенз-альдегида путем диазореакции | Бесцветные кристаллы; 1°пл. Ю8°; t°icun· 240° | В производстве три-фенилметановых красителей (патентованный голубой)
• |
| Тиосалициловая кислота | Из антранило-вой кислоты путем диазореакции | Желтые кристаллы 1°пл. 1^3° | Для производства тилонафтеновых кубовых красителей | |
| Дегидротио-п-толуи-
дин |
C-<Z>--NH3
/V. СНз ь |
Нагреванием n-толуидина с серой при высокой ί° (140—220°) | Нерастворим в воде; (°И4. 191°; 1°кип· 131° | Получение приму-лина и тиофлавина |
| Пикраминовая кислота (динитро-о-аминофенол) | он
N0,-0— ΝΗο 1 NO 2 |
Восстановлением пикриновой кислоты | Красные кристаллы; 1°пл, 169° | Для хромирующихся красителей |
| Бензотрихлорид | /ОСЬ
0 |
Хлорированием бензола | Бесцветная жидкость; 1°пл. 21,2°; ί°ηϊΐγι. 214°; П14 1,380 | Для получения зеленого малахитового (при взаимодействии с димотил-анилкном) |
| 1,5-Динитронафта-
лин |
NO 2
CO NO 2 |
Нитрованием нафталина | Кристаллы слабо желтого цвета; t°nA. 215° | Для нафтазарина |
Схема реакций получения П. п. Все приведенные реакции взаимно связаны друг с другом и генетически, что м. б. изображено следующей схемой (для удобства взят бензол и простейшие его производные; пунктиром отмечены отдельные ответвления общей реакции)
ксимальные количества 1,8-изомера по схеме
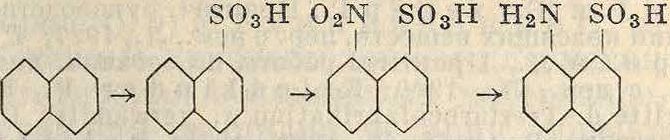
Если мы проведем реакции в иной последовательности, именно нитрование, сульфиро-
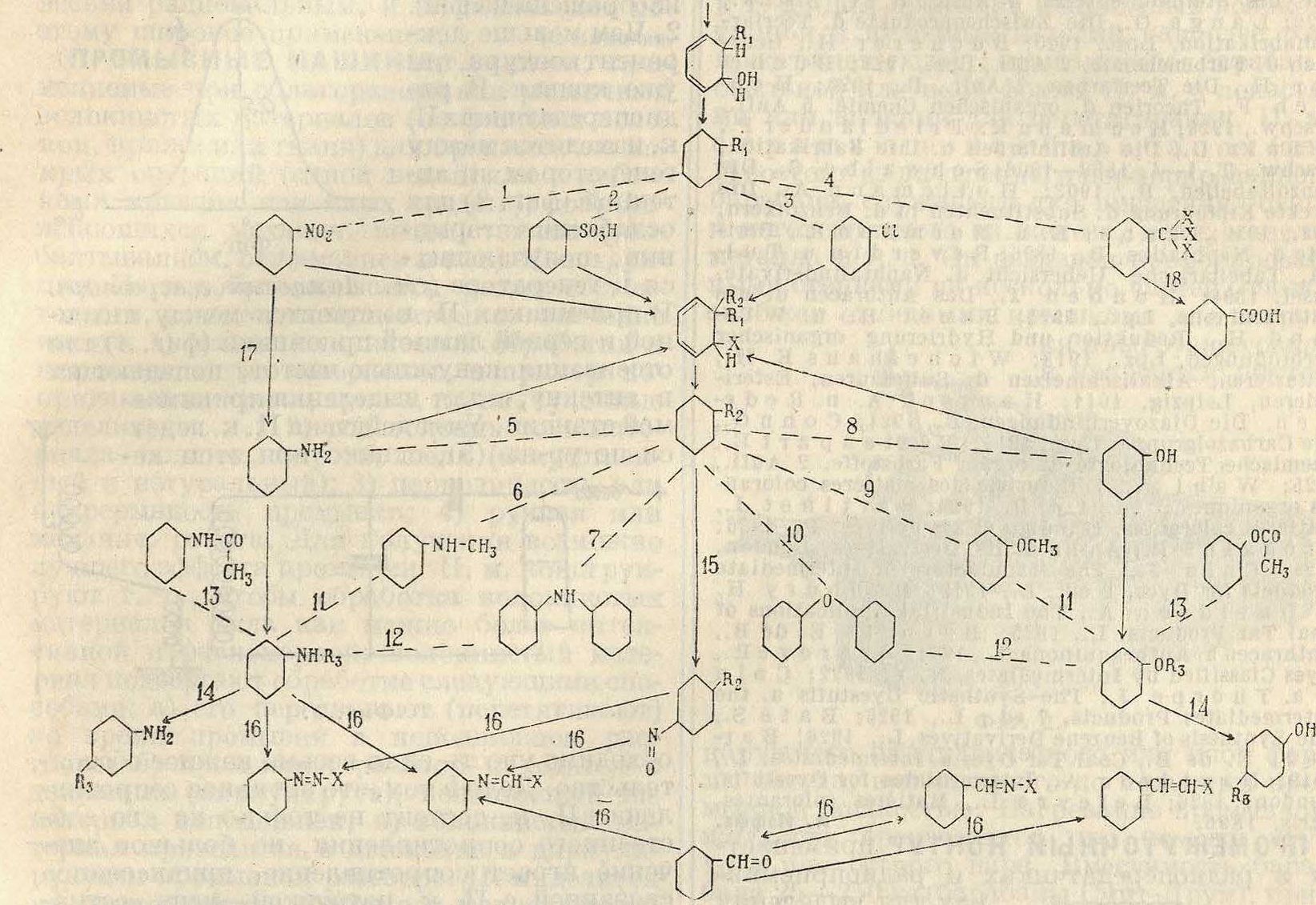
На схеме линиями обозначены реакции: ί— введения нитрогруппы, 2—введения суль-фогруппы, 5—введения галоида, 4—введения углеродсодержащей группы, 5—амини-рования, 6—алкаминирования, 7—арамини-ровамм, 8—гидроксилирования, 9—алк-окоилирования, 10—ароксилирования, 11— алкилирования, 12—арилирования,! 3—ацилирования, 14—перегруппировки, 15—ни-трозирования, 16—введения двувалентных групп, 17—восстановления, 18—окисления.
Как видно из вышеизложенного, П. п. в большинстве получаются в результате ряда последовательных операций, и необходимо уметь эти операции провести в необходимой наиболее удобной последовательности, т. к. порядок реакций весьма существенно влияет как на технологич. ведение процесса, так и на строение конечного продукта. Для примера разберем получение сульфокислоты а-нафтиламина. Исходя из нафталина, мы должны провести реакции нитрования, восстановления и сульфирования, причем порядок их играет весьма большую роль. Естественно, что операция восстановления должна следовать за операцией нитрования, но реакция сульфирования м. б. проведена в начале, в середине и в конце. В первом случае мы имеем последовательно реакции; сульфирования, нитрования и восстановления, и в конечном итоге получим, в силу ориентирующего действия сульфогруппы.ма-
вание и восстановление, то доминирующим изомером у нас будет 1,5-, наряду с нек-рым количеством 1,8-изомера. Технологически у нас процесс будет сложнее как в силу необходимости заранее высушить нитропродукт, так и вследствие более трудного сульфирования нитронафталина. Схема процесса для главного изомера следующая
ΝΟϋ no2 NHS
I I
S03H S03H
Если реакцию сульфирования проведем в конце, то главным продуктом реакции будет 1,4- изомер, наибольшее количество которого будет получаться при ведении бак-процесса по схеме
ΝΟϋ NHs NHa i i [
S03H
Из сказанного очевидно, что при получении сложных П. и. большую роль играют способ их получения и выбор варианта реакции.
Лит.: ВорожцовИ.Н., Основы синтеза красителей, М.—Л., 1925; его те, Ступени в синтезе красителей, Л., 1926; Чичибабин А.Е., Основные начала органич. химии, 3 изд., М.—Л., 1931; Залькинд Ю. С., Химия циклич. соединений, 2 изд., Л., 1930; Ф и р ц-Д а в и д Г., Производство органич. красок, пер. с нем., М.—Л., 1927; М е-лау Р. и Бухер ер Г., Практич. руководство по химии красящих веществ, пер. с нем., Л., 1927; Раттер м а н Л., Практич. работы по органич. химии, пер. с нем., Л., 1926; Friedlander Р., Fort-schritte d. Teerfarbenfabrikation u. verwandter Indu-striezweige, В. 1—16, B., 1891—1928; Beilstein F., Handbuch d. organischen Chemie, 4 Aufl., В. 1—5, B., 1918— 22; Mayer V. u. Jacobson P., Lehrbuch d. organischen Chemie, 2 Aufl., B. 2, T. 1—2, 1923; Η о u b e n-W e у 1, Die Metboden d. organi-schen Chemie, B. 2, Lpz., 1925; SchulzS., Die Chemie des Steinkohlenteers, 4 Aufl., В. 1, Brsehw., 1926; Lange O., Die Zwischenprodukte d. Teerfar-benfabrikation, Lpz., 1920; Bucherer H., Lehrbuch d. Farbenchemie, 2 Aufl., Lpz., 1921; Bucherer H., Die Teerfarben, 2 Aufl., B., 1920; Hen-rich F., Theorien d. organischen Chemie, 5 Aufl., Brsehw., 1924; HeumannK., 1’riedlanderP., Schulz G., Die Anilinfarben u. ihre Fabrikation, Brsehw., T. 1—4, 1888—1906; Schwalbe C., Die Benzoltabellen, B., 1903; Hollemann A., Die direkte Einfiihrung d. Substituenten in d. Benzolkern, Lpz., 1910; Tauber E. u. Normann R., Derivate d. Naphtalins, B., 1896; R e у e r d i n u. T u 1-d a, Tabellarische tfebersicht d. Naphtalinderivate, BasPl, 1894; H oub en J., Das Anthracen u. die Anthrachinone, Lpz., 1928; Bauer R. u. W i e-land H., Reduktion und Hydrierung organischer Verbindungen, Lpz., 1918; W i c h e 1 h a u s К. H., Sulfurieren, Alkalischmelzen d. Sulfosauren, Esterl-fizieren, Leipzig, 1911: Hantsch A. u.Sede-lien, Die Diazoverbindungen, B., 1921; С о h n G., Die Carbazolgruppe, Lpz., 1919; Ristenpart E., Chemische Technologies, organ. Farbstoffe, 2 Aufl., 1925; Wahl A., L’industrie des matlires colorantes organiques, 2 6d., t. 1, P., 1926; Martinet I., MatiOres colorantes, L’indigo et ses dOrivds, P., 1926; G r о g g i n s P., Anilin a. its Derivatives, London, 1924; Cain J., The Manufacture of Intermediate Products for Dyes, 2 ed., L., 1919; B u n b u г у H, a. Davidson A., The Industrial Applications of Coal Tar Products, L., 1925; Barnett E. de B., Anthracen a. Anthraquinone, L., 1921; S e h r e v e R„ Dyes Classified by Intermediates, N. Y., 1922; Cain
J. a. Thorpe J., The Synthetic Dyestuffs a. the Intermediates Products, 7 ed., L., 1926; Bate S., The Synthesis of Benzene Derivatives, L., 1926; Barnett E. de B., Coal Tar Dyes a. Intermediates, L., 1919; Davidson A., Intermediates for Dyestuffs, London, 1926; Lefevre L., Matiferes colorames, Paris, 1896. И. Иоффе.