 > Техника, страница 96 > Спектральный анализ
> Техника, страница 96 > Спектральный анализ
Спектральный анализ
Спектральный анализ (при помощи спектров испускания) имеет применение почти во всех отраслях х-ва. Широко применяется в металлопромышленности для быстрого анализа железа, стали, чугуна, а также различных специальных сталей и готовых металлич. изделий, для установления чистоты легких, цветных и драгоценных металлов. Большое применение имеет С. а. в геохимии при изучении состава полезных ископаемых. В химической нром-сти и близких к ней отраслях С. а. служит для установления чистоты выпускаемой и применяемой продукции, для анализа катализаторов, различных остатков, осадков, мутей и промывных вод; в медицине — для открытия металлов в различных органических тканях. Ряд специальных задач, трудно разрешаемых или вовсе не разрешимых иным путем, решается при помощи С. а. быстро и точно. Сюда относится например распределение металлов в сплавах, исследование в сплавах и минералах сульфидных и других включений; такого рода исследования иногда обозначаются термином локальный анализ. Выбор того или другого типа спектрального аппарата сточки зрения достаточности его дисперсии производится в зависимости от цели и задач С. а. Для исследования платиновых металлов (Ru, Rh, Pd, Os, Ir, Pt), а также Fe, Co, Ni, Сг, V, Mo, W, Ti, Mn, Zr, Re, Nb и Та наиболее пригодны кварцевые спектрографы с большей дисперсией, дающие для длин волн 4 000—2 200 А полоску спектра длиной по крайней мере 22 см. Для остальных элементов м. б. применены аппараты, дающие спектры длиной 7—15 см. Спектрографы со стеклянной оптикой в общем имеют меньшее значение. Из них удобны комбинированные приборы (например фирмы Гильгера и Фюсса), которые по желанию можно применять в качестве спектроскопа (смотрите) и спектрографа (смотрите). Для получения спектров применяются следующие источники энергии. 1) Пламя горящей смеси— водорода и кислорода, смеси кислорода и светильного газа, смеси кислорода и ацетилена или наконец воздуха и ацетилена I1]. В последнем случае t° источника света доходит до 2 500—3 000°. Пламя наиболее всего пригодно для получения спектров щелочных и щелочноземельных металлов, а также для таких элементов, как Си, Hg и Т1. 2) Вольтовадуга. а) Обычная, гл. обр. постоянного тока, силой
5—20 А. С большим успехом она применяется для качественного анализа трудно сплавляемых минералов, которые вводятся в дугу в виде кусочков или тонко растертых порошков. Для коли чественного анализа металлов применение обычной вольтовой дуги имеет очень существенный недостаток, заключающийся в том, что поверхность анализируемых металлов покрывается пленкой окиси и горение дуги становится в конце-концов неравномерным. Темп-pa вольтовой дуги доходит до 5 000—6 000°. б) Прерывистая дуга (Abreissbogen) [2,4] постоянного тока силой 2— 5 А при напряжении ок. 80 V. При помощи специального приспособления горение дуги прерывается 4—10 раз в ск. Этот способ возбуждения уменьшает окисление поверхности анализируемых металлов. При более высоком напряжении — до 220 Y и силе тока 1—2 А— прерывистая дуга может применяться также и для ана-
LF
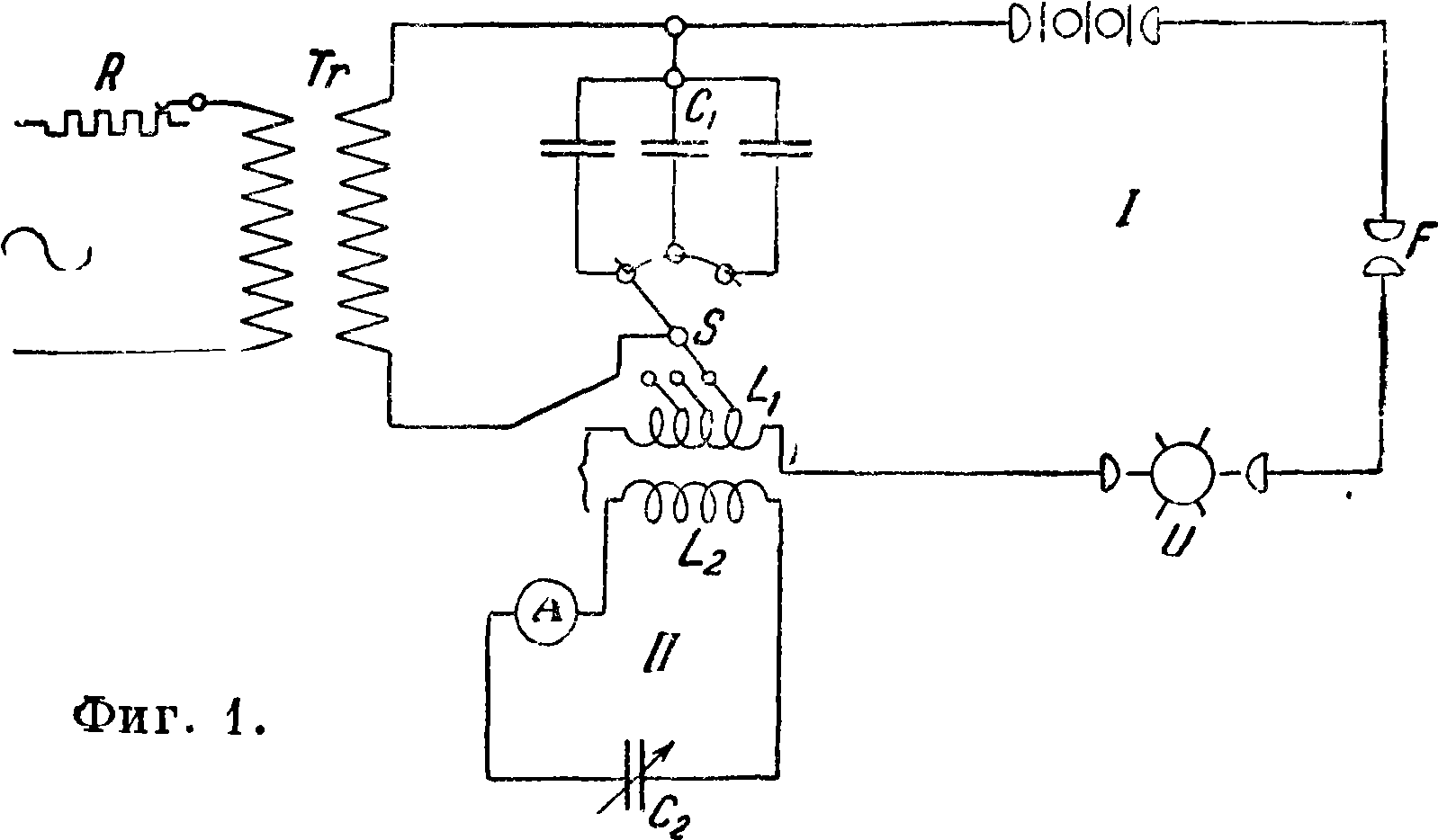
лиза растворов. 3) Искровые разряды, получаемые при помощи индукционной катушки или, чаще, трансформатора постоянного или (предпочтительнее) переменного тока мощностью до 1 kW, дающего во вторичной цепи 10 000— 30 000 V. Применяются три типа разрядов, а) Искровые разряды без емкости и индуктивности во вторичной цепи, называемые иногда дугой высокого напряжения (Hochspannungsbogen). Анализ жидкостей и расплавленных солей при помощи таких разрядов отличается большой чувствительностью. б) Искровые разряды с емкостью и индуктивностью во вторичной цепи, часто называемые также конденсированными искрами, представляют собой более универсальный источник энергии, пригодный для возбуждения спектров почти всех элементов (кроме щелочных металлов), а также газов. Схема включения дана на фигуре 1, где R — реостат в первичной цепи, Тг — трансформатор переменного тока, Сг—емкость во вторичной цепи /, S — переключатель для изменения индуктивности Llt U — синхронный прерыватель, LF —
искрогаситель, F— рабочий искровой промежуток. В резонанс ко вторичной цепи I при помощи индуктивности L2 и переменной емкости С2 настраивается вторичная цепь //; признаком наличия резонанса является наибольшая сила тока, показываемая миллиамперметром А. Назначение вторичной цепи II синхронного прерывателя U и искрогасителя LF — делать электрические разряды возможно однообразными как по характеру, так и по числу в течение определенного промежутка времени; при обычных работах такие добавочные приспособления не вводятся. При исследованиях металлов во вторичной цепи при
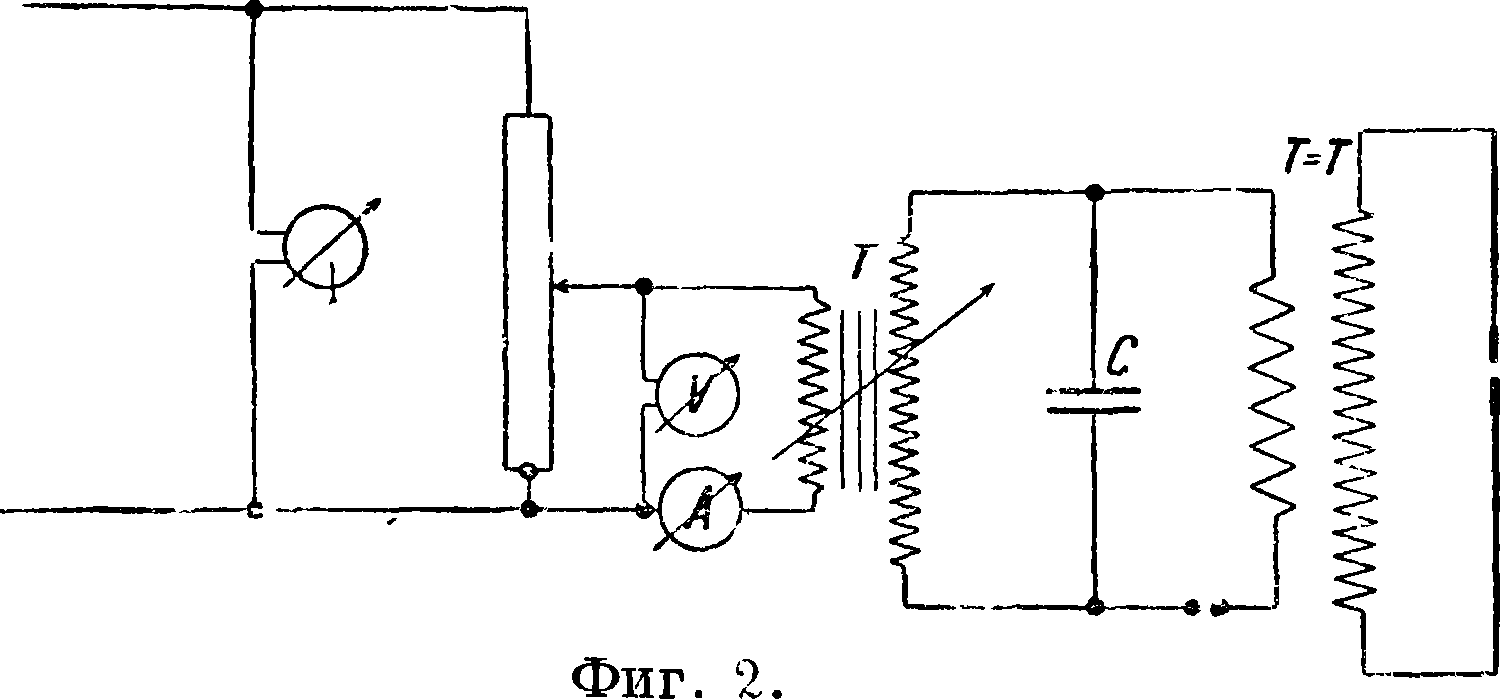
меняется ёмкость 6 000—15 000 сантиметров и индуктивность до 0,05—0,01 Н. Для анализа жидкостей во вторичную цепь иногда вводится водяной реостат с сопротивлением до 40 000 Ω. Газы исследуются без индуктивности с небольшой емкостью. в) Разряды токов Тесла, которые осуществляются при помощи схемы, изображенной на фигуре 2, где V — вольтметр, А—амперметр,
Фигура 3.
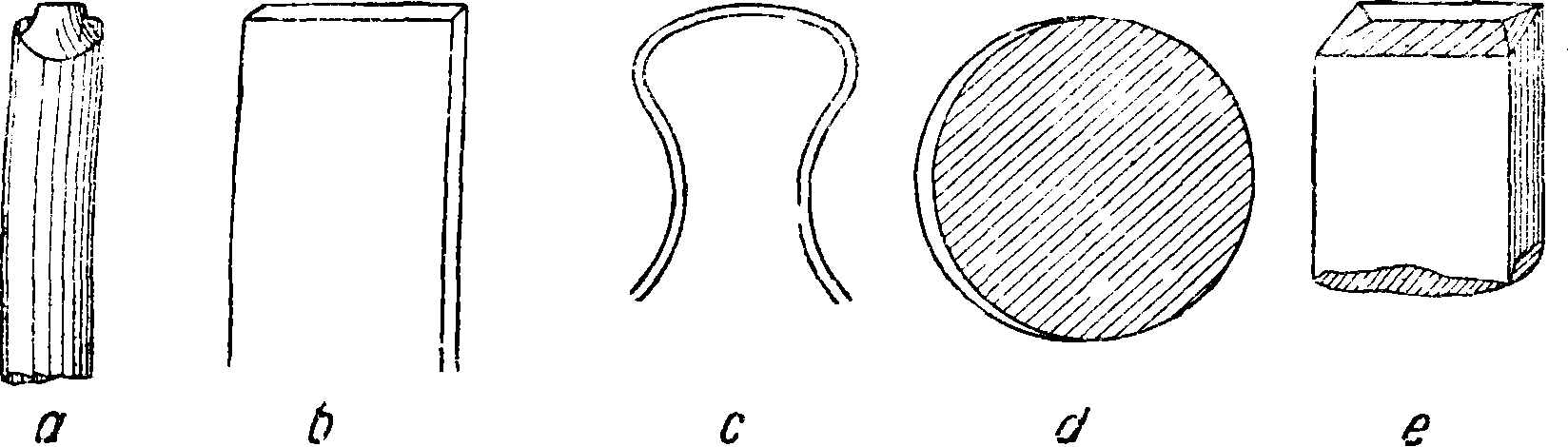
Т — трансформатор, С — емкость, Т—Т—трансформатор Тесла, F — искровой промежуток, куда вводится анализируемое вещество. Токи Тесла применяются для исследований веществ, которые имеют невысокую точку плавления: различных растительных и органич. препаратов, осадков на фильтрах и тому подобное. При С. а. металлов в случае большого их количества они обычно сами являются электродами, причем им придается какая-либо форма, например из указанных на фигуре 3, где а — электрод из анализируемой толстой проволоки, b—из жести, с — со
Фигура 4.
Фигура 5.
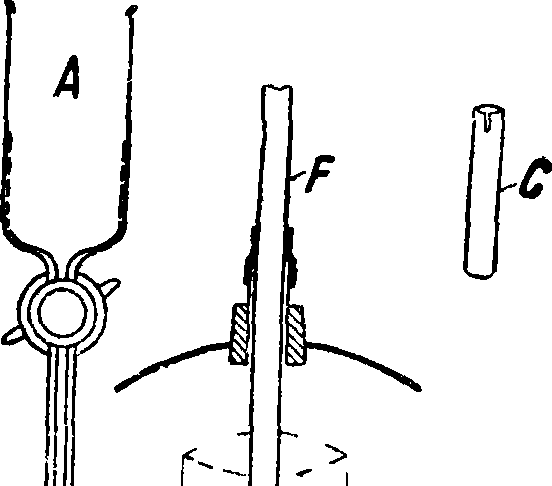
гнутая тонкая проволока, d—диск, отрезанный от толстого циликдрич. стержня, е—форма, выпиливаемая из больших кусков литья. При количественном анализе необходимо иметь всегда одинаковую форму и размеры подвергающейся действию искр поверхности электродов. При небольшом количестве анализируемого металла можно воспользоваться оправой из какого-либо чистого металла, например из золо!а и платины, в которой укрепляется анализируемый металл, как показано на фигуре 4. Для введения в источник света растворов предложено довольно много способов. При работе с пламенем применяется распылитель
Люндегорда [“,2], схематически изображенный на фигуре 5 вместе со специальной горелкой. Продуваемый через распылитель ВС воздух захватывает испытуемую жидкость, наливаемую в количестве 3 —10 см3 в углубление С, и в виде тонкой пыли относит ее в горелку А, где происходит смешение с газом. Для введения растворов в дугу, а также в искру применяются чистые угольные или графитовые электроды, на одном из которых делается углубление. Необходимо однако отметить, что очень трудно приготовить угли совершенно чистыми. Применяемые для очистки способы — попеременное кипячение в соляной и плавиковой кислотах, а также прокаливание в атмосфере водорода до 2 500—3 000°— не дают углей, свободных от примесей, остаются (хотя и следы) Са, Mg, V, Ti, AI, Fe, Si, В. Удовлетворительной чистоты получаются также угли путем прокаливания их на воздухе при помощи электрич. тока: через угольный стержень диам. 5 миллиметров пропускается ток силой ок. 400 А, и достигаемое таким путем сильное накаливание (до 3 000°) оказывается достаточным для того, чтобы в течение нескольких секунд большинство загрязняющих угли примесей уле
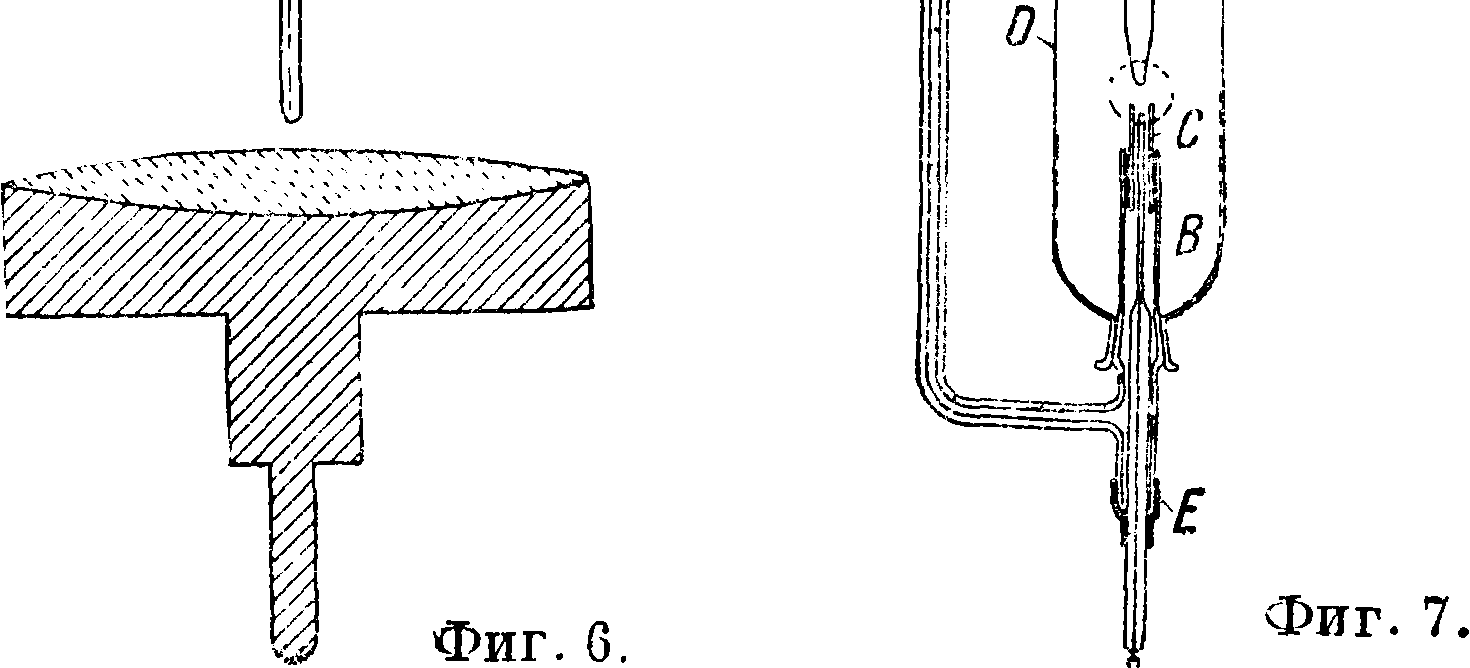
тучилось [10]. Существуют также такие способы введения растворов в искру, где сам раствор является нижним электродом и искра проскакивает на его поверхность; другим электродом может служить какой-либо чистый металл. Примером такого устройства может служить изображенный на фигуре 6 жидкостный электрод Герляха [3]. Углубление, куда наливается испытуемый раствор, облицовывается платиновой фольгой или покрывается толстым слоем позолоты. На фигуре 7 изображен аппарат Хитчена, служащий также для введения растворов в искру. Из сосуда А испытуемый раствор слабой струей поступает через трубку В и кварцевую насадку С в сферу действия искровых разрядов. Нижний электрод, впаянный в стеклянную трубку, прикрепляется к аппарату при помощи каучуковой трубки Е. Насадка С, изображенная на фиГ. 7 отдельно, имеет с одной стороны вырез для стенания раствора. D—стеклянный предохранительный сосуд, в котором делается круглое отверстие для выхода ультрафиолетовых лучей. Сосуд этот удобнее делать кварцевым без отверстия. К верхнему электроду F, графитовому, угольному или металлическому,^ также приспосабливается предохраняющая от брызг пластинка. Для «дуги высокого напряжения», сильно накаливающей анализируемые вещества, Герлях [4] при работе с растворами применяет электроды с охлаждением, как это схематичес-
*2Я
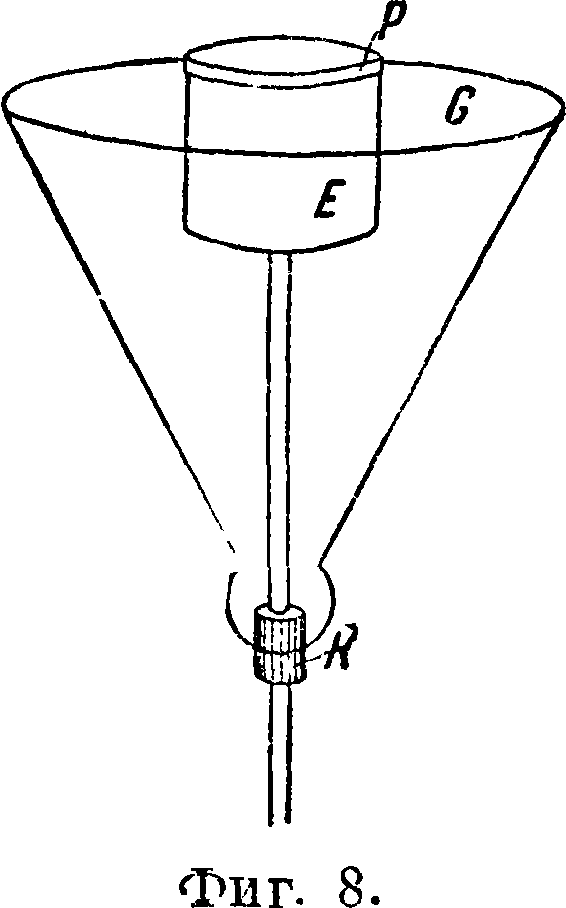
ни показано на фигуре 8. На толстой проволоке (диам. 6 миллиметров) укрепляется при помощи пробки К стеклянная воронка G, куда помещаются кусочки льда. На верхнем конце проволоки
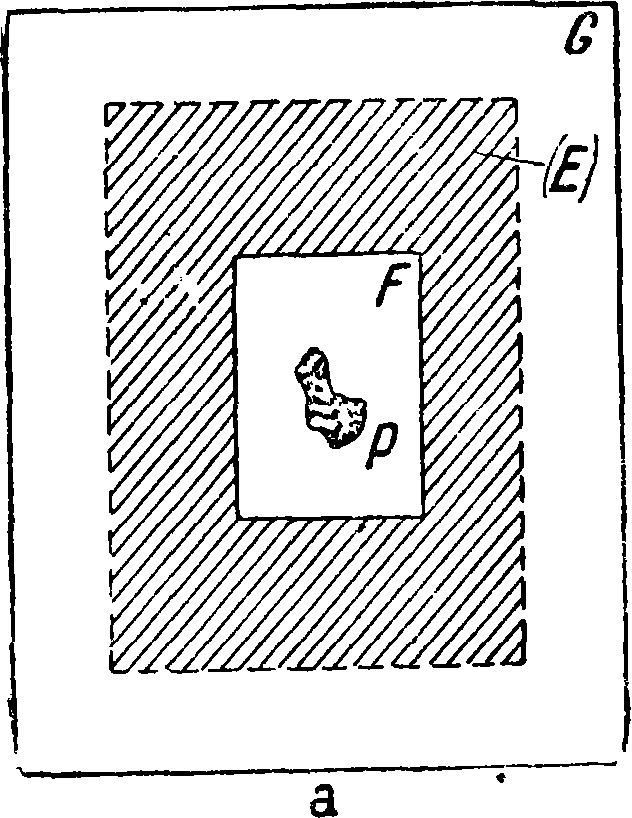
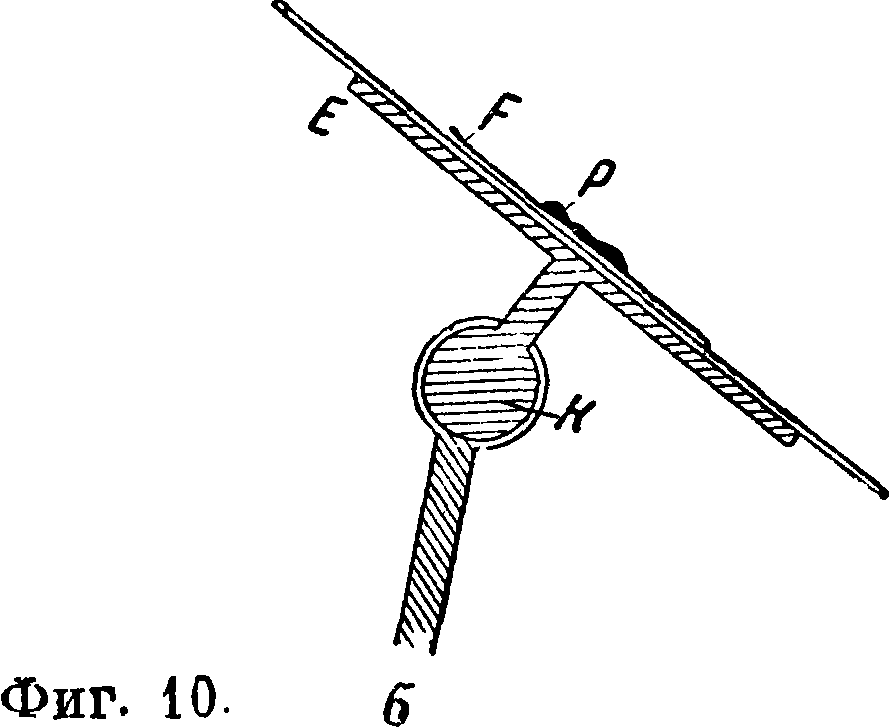
S укрепляется круглый железный электрод Е диам. 4 сантиметров и высотой 4 см, на к-рый накладывается платиновая чашечка Р; последняя должна легко сниматься для очистки. Верхний электрод также должен быть толстым во избежание расплавления. При анализе небольших количеств веществ—осадков на фильтрах, различных порошков и т. д.— можно пользоваться приспособлением, изображенным на фигуре 9. Из испытуемого вещества и фильтровальной бумаги делается комо-— чек, смачивается для лучшей проводи-Фигура 9. мости раствором например NaCl, помещается на нижний электрод, состоящий иногда из чистого кадмия, заключенного в кварцевой (хуже стеклянной) трубочке; верхний электрод также является каким-либо чистым металлом. Для таких же анализов при работе с токами Тесла применяется специальная конструкция искрового промежутка [4], изображенная на фигуре 10 а и б.
В круглом шарнире К укрепляется в нужном положении алюминиевая пластинка Е, на к-рую накладывается стеклянная пластинка G, а на последнюю—препарат Р на фильтровальной бумаге^. Препарат смачивается какой-либо к-той или раствором соли. Вся эта система представляет небольшой конденсатор. Для исследования газов применяются закрытые стеклянные или кварцевые сосуды (фигура 11).
Для количественного анализа газов удобно пользоваться золотыми или платиновыми электродами, линии которых можно применить для сравнения.
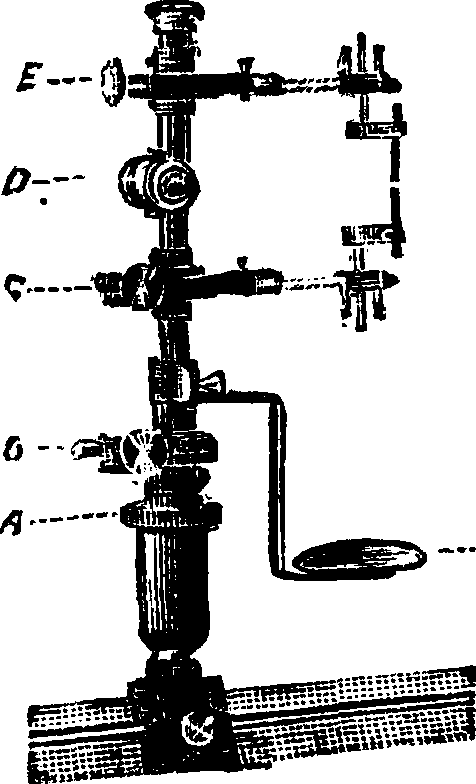
Почти все из упомянутых выше приспособлений для введения веществ в искру и дугу при работе укрепляются в специальных штативах. Примером может являться штатив Гра-мона, изображенный на фигуре 12: при помощи винта D электроды одновременно раздвигаются и сдвигаются; винт Е служит для передвигания верхнего электрода параллельно оптич. скамье, а винт С—для боковых поворотов нижнего электрода; для боковых поворотов всей верхней части штатива служит винт В; наконец при помощи винта А можно поднимать или опускать всю верхнюю часть штатива; Н — подставка для горелок, стаканов и прочие Выбор источника энергии для той или иной цели исследования можно сделать, руководствуясь следующей примерной таблицей.
Качественный анализ. При качественном С. а. открытие какого-либо элемента зависит от многих факторов: от характера определяемого элемента, источника энергии, раз
решающей способности спектрального аппарата, а также от чувствительности фотографических пластинок. Относительно чувствительности анализа можно сделать следующие указания. При работе с искровыми разрядами в растворах
| Характер пробы | Источник энергии J | Цель исследования |
| Металлы, большие количества. Готовые ме-таллич. изделия | 1. Искровые «конденсированные» разряды
2. Вольтова дуга: а) обычная б) прерывистая |
Качественный и количественный анализы
Качественный анализ Количественный анализ |
| Металлы, небольшие количества | 1. Искровые «конденсированные» разряды
2. Токи Тесла |
Качественный и количественный анализы |
| Тонкая шесть или проволока | 1. Искровые «конденсированные» разряды
2. Токи Тесла |
Качественный и количественный анализы |
| Минералы в кусках или порошках, сплавленные | 1. «Дуга высокого напряжения»
?. Токи Тесла 3. Вольтова дуга обычная |
j Качественный анализ |
| Минералы и другие вещества в порошках | 1. «Дуга высокого напряжения»
2. Токи Тесла 3. Вольтова дуга обычная 4. Пламя по Люндегорду |
1 Качественный и количе-1 ственный анализы |
| Жидкости, большие количества | 1. «Дуга высокого напряжения»
2. Искровые «конденсированные» разряды с электродом Герляха, аппаратом Хитчена 3. Пламя по Люндегорду |
Качественный анализ
1 Качественный и коли-I чественный анализы |
| Жидкости, небольшие количества (0,5—J,Осле3) | 1. Токи Тесла со смоченным и высушенным фильтром
2. Искровые «конденсированные» разряды с угольными или графитовьши электродами 3. Вольтова дуга обычная с теми же электродами |
Качественный и количественный анализы |
| Осадки на фильтрах | Токи Тесла | Качественный анализ |
| Газы | Искровые «конденсированные» разряды | Качественный и количественный анализы |
можно открывать 10 9—10 3%, а в мет 10“2—10“4 % исследуемого элемента; при работе с вольтовой дугой пределы открытия лежат около 10“3%. Абсолютное количество, которое м. б. открыто при работе с пламенем, составляет 10“4—10“ 7 г, а при искровых разрядах 10“6— 10“8 г исследуемого элемента. Наибольшая чув-
ствительность открытия относится к металлам и металлоидам—В, Р, С; меньше чувствительность для металлоидов As, Se и Те; галоиды, а также S, О, N в их соединениях совсем не м. б. открыты и м. б. открыты лишь в некоторых случаях в газовых смесях. Для качественного анализа наибольшее значение имеют «последние линии», и при анализе задача заключается в наиболее точном определении длин волн спектральных линий. При визуальных исследованиях длины
Фигура и.
Фигура 12.
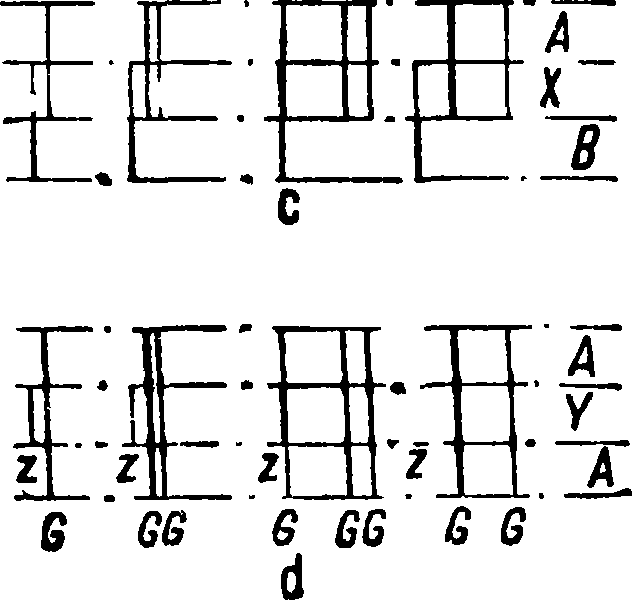
волн отсчитываются по барабану спектрометра; эти измерения можно считать лишь приблизительными, так как точность составляет обычно +2—ЗА и в таблицах Кайзера [45] этому интервалу ошибок могут отвечать ок. 10 спектральных линий, принадлежащих различным элементам, для Я 6 000 и 5 000 А и ок. 20 спектральных линий для Я ^ 4 000 А. Гораздо точнее определяется длина волн при спектрографич. анализе. В этом случае на спектрограммах при помощи измерительного микроскопа измеряется расстояние между линиями с известной длиной волны и определяемой; по формуле Гартмана находится длина волны последней. Точность таких измерений при работе с прибором, дающим полоску спектра длиной ок. 20 см, составляет i 0,5 А для Я ^ 4000 А, ± 0,2 А для Я ^ 3 000 А и ^ 0,1 А для Я ^ 2 500 А. По длине волны в таблицах находят соответствующий элемент. Расстояние между линиями при обычных работах измеряется с точностью до 0,05 — 0,01 миллиметров. Этот прием иногда удобно комбинировать со съемками спектров с так называемым заслонками Гартмана, два типа которых изображены на фигуре 13, а и Ь; при помощи их щель спектрографа можно делать различной высоты. Фигура 13, с схематически изображает случай качественного анализа вещества×— установление в нем элементов А и В. Спектры фигура 13, d показывают, что в веществе Y кроме элемента А, линии которого обозначены буквой G, имеется примесь, линии которой обозначены Z. При помощи этого приема в простых случаях можно выполнить качественный анализ, не прибегая к промеру расстояний между линиями.
Количественный анализ. Для количественного С. а. наибольшее значение имеют линии, обладающие возможно большей концентрационной чувствительностью где — ин тенсивность линии, а К — концентрация дающего ее элемента. Чем больше концентрационная чувствительность, тем точнее анализ. С тече
Фигура 13.
нием времени разработан целый ряд методов количественного С. а. Эти методы следующие.
I. Спектроскопические методы (без фотографии, съемки) почти все являются фотометрия. методами. Сюда относятся: 1) Метод Барратта [п]. Одновременно возбуждаются спектры двух веществ—испытуемого и стандартного,—видные в поле зрения спектроскопа рядом, один над другим. Ход лучей изображен на фигуре 14, где Fx и F2 — два искровых промежутка, свет от которых проходит через призмы Николя и JV2, поляризующие лучи во взаимно перпендикулярных плоскостях. При помощи призмы D лучи попадают в щель S спектроскопа. В его зрительной трубе помещается третья призма Николя—анализатор,—вращая к-рую добива
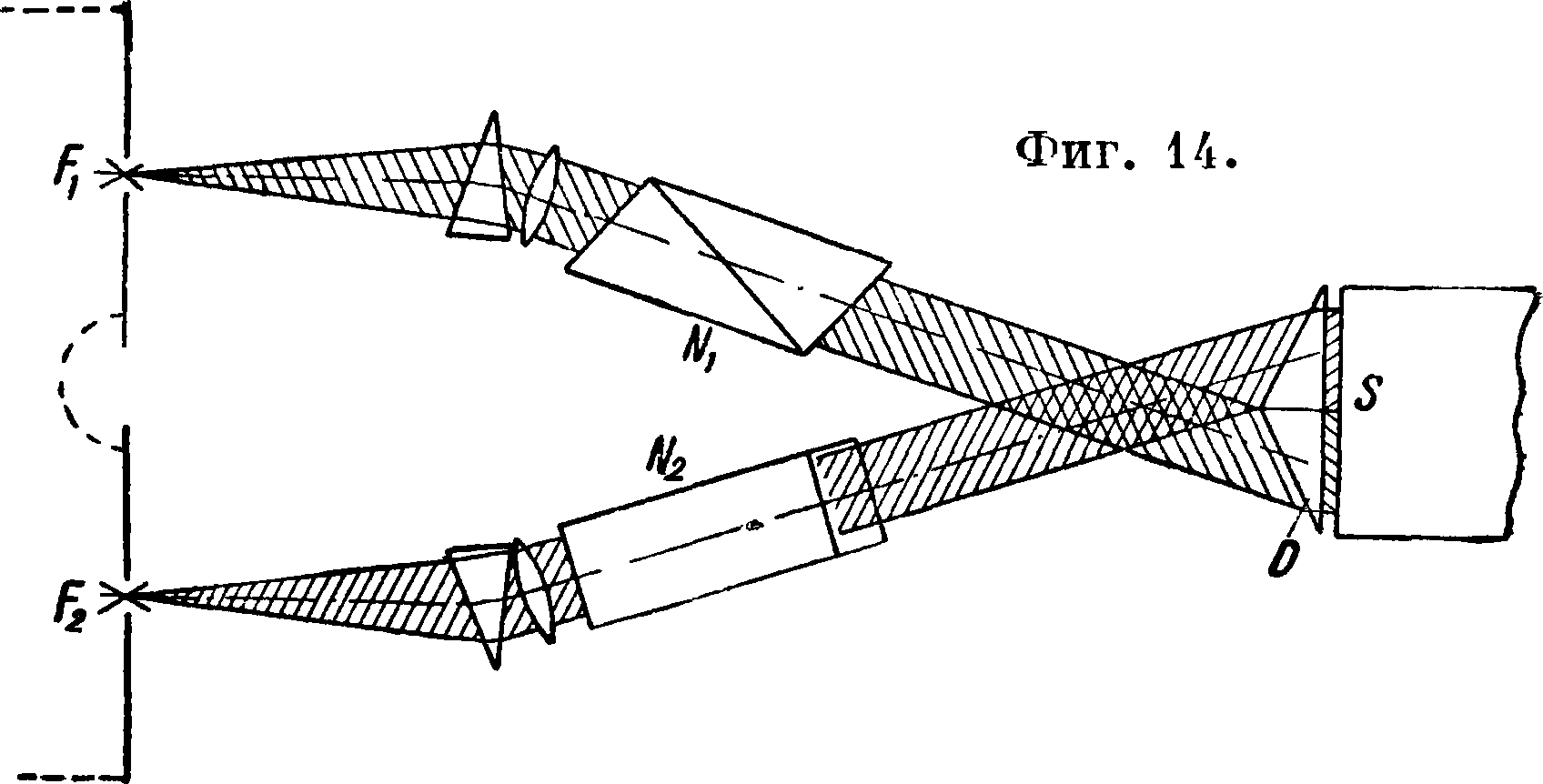
ются одинаковой интенсивности двух сравниваемых линий. Предварительно при исследованиях стандартов, то есть веществ с известным содержанием элементов, устанавливается зависимость между углом поворота анализатора и концентрацией, и по этим данным вычерчивается диаграмма. При анализе по углу поворота анализатора из этой диаграммы находится искомое процентное содержание. Точность метода ±10%. 2) М е-
тод Шейбе и Лиммера [12,13,14]. Принцип метода заключается в том, что лучи света
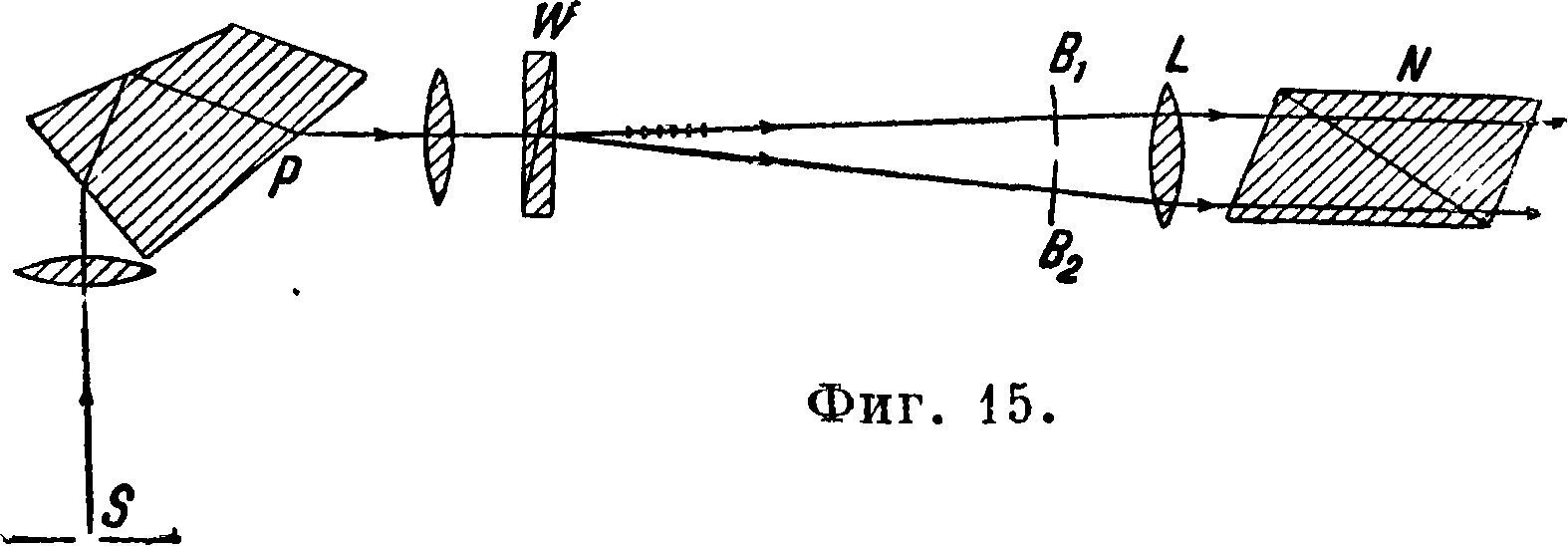
после призмы спектроскопа проходят через призму Волластона, где расходятся на два пучка и поляризуются во взаимно перпендикулярных плоскостях. Схема хода лучей показана на фигуре 15, где S — щель, Р — призма спектроскопа, W — призма Волластона. В поле зрения получаются два спектра Вх и В2, лежащие рядом, друг над другом; L — лупа, N — анализатор. Если вращать призму Волластона, то спектры будут передвигаться относительно друг друга, что позволяет совместить какие-либо две их линии. Например если анализируется железо, содержащее ванадий, то совмещается линия ванадия с какой-либо близлежащей одноцветной линией железа; затем, поворачивая анализатор, добиваются одинаковой яркости этих линий. Угол поворота анализатора, как и в предыдущем методе, является мерой концентрации искомого-элемента. Метод особенно пригоден для анализа железа, спектр которого имеет много линий, что позволяет всегда найти линии, пригодные для исследований. Точность метода ±3—7%. 3) М е-тод Оккиалини [15,16]. Если расположить электроды (например анализируемые металлы) горизонтально и проектировать изображе! иэ ис точника света на вертикальную щель спектроскопа, то как при искровых, так и при дуговых разрядах линии примесей м. б. открыты в зависимости от концентрации на большем или меньшем расстоянии от электродов. Источник света проектируется на щель при помощи специальной линзы, снабженной микрометрическим винтом. При анализе эта линза передвигается и вместе с ней передвигается изображение источника света до тех пор, пока какая-либо линия примеси в спектре исчезнет. Мерой концентрации примеси является отсчет по шкале линзы. В настоящее время этот метод разработан также и для работ с ультрафиолетовой частью спектра. Надо отметить, что таким же способом освещения щели спектрального аппарата пользовался Локиер [17] и им был разработан метод количественного G. а., т. н. метод «длинных и коротких линий».
4) Прямое фотометрирование спектров. Описанные выше методы носят название визуальных. Люндегорд р,2,18] вместо визуальных исследований пользовался для измерения интенсивности спектральных линий фотоэлементом. Точность определения щелочных металлов при работе с пламенем достигала +5%. При искровых разрядах этот способ неприменим, так как они менее постоянны, чем пламя. Существуют также способы, основанные на изменении индуктивности во вторичной цепи [19], а также использующие искусственное ослабление света, попадающего в „спектроскоп, до исчезновения в поле зрения исследуемых спектральных линий [20].
II. Спектрографические мето-д ы. При этих методах исследуются фотография, снимки спектров, причем мерой интенсивности спектральных линий является почернение, даваемое ими на фотография, пластинке. Интенсивность оценивается или глазом или фотометрически. А. Методы без применения фотометрии. 1) Метод последних линий [21~31]. При изменении концентрации какого-либо элемента в спектре изменяется число его линий, что дает возможность при неизменных условиях работы судить о концентрации определяемого элемента. Фотографируется ряд спектров веществ с известным содержанием интересующего компонента, на спектрограммах определяется число его линий и составляются таблицы, в которых указывается, какие линии видны при данных концентрациях. Эти таблицы служат дальше для аналитич. определений. При анализе на спектрограмме определяется число линий интересующего элемента и по таблицам находится процентное содержание, причем метод дает не однозначную его цифру, а границы концентраций, то есть «от—до». Наиболее достоверно возможно различить концентрации, отличающиеся друг от друга в 10 раз, например от 0,001 до 0,01%, от 0,01 до 0,1% и т. д. Аналитич. таблицы имеют значение лишь для вполне определенных условий работы, которые в различных лабораториях могут очень сильно различаться; кроме того требуется тщательное соблюдение постоянства условий работы. 2) Метод сравнительных спектров [32“36]. Фотографируется несколько спектров анализируемого вещества А + х% В, в к-ром определяется содержание х элемента В, и в промежутках между ними на той же фотография, пластинке—спектры стандартных веществ А + а%В, А + b% В, А + с% В, где а, Ь, с — известное процентное содержание В. На спектрограммах по интенсивности линий В определяется, между какими концентрациями заключается значение х. Критерием постоянства условий работы является равенство интенсивности на всех спектрограммах какой-либо близлежащей линии А. При анализе растворов в них добавляется одинаковое количество какого-либо элемента, дающего линию близко к линиям В, и тогда о постоянстве условий работы судят по равенству интенсивности этих линий. Чем меньше разница между концентрациями а,b,сл. и чем точнее достигнуто равенство интенсивности линий А, тем точнее анализ. А. Райс [ 33] например применял концентрации а, Ь, с,., относящиеся друг к другу, как 1 : 1,5. К методу сравнительных спектров примыкает метод «подбора концентраций» (Test-verfahren) по Гюттигу и Турнвальду [37], применимый только к анализу растворов. Он заключается в том, что если в двух растворах, содержащих а% А и х% А (х^= а), что сейчас же можно определить по их спектрам, то прибавляют в какой-либо из этих растворов такое количество п элемента А, чтобы интенсивность его линий на обоих спектрах стала одинаковой. Тем самым определится концентрация х, которая будет равна (а + п)%. Можно также прибавить в анализируемый раствор какой-либо другой элемент В до равенства интенсивности определенных линий А и В и по количеству В оценить содержание А. 3) Метод гомологических пар [зэ]. В спектре вещества А + а% В линии элементов А и В не являются одинаково интенсивными и, если этих линий достаточное количество, можно найти две такие линии А и В, интенсивность которых будет одинакова. Для другого состава А + b% В одинаковыми по интенсивности будут другие линии А и В и т. д. Эти две одинаковые линии называются гомологическими парами. Концентрации В, при которых осуществляется та или иная гомология, пара, называются фиксирующими пунктами этой пары. Для работы по этому методу требуется предварительное составление таблиц гомология, пар при помощи веществ известного состава. Чем полнее таблицы, то есть чем больше они содержат гомология, пар с фиксирующими пунктами, отличающимися как можно меньше друг от друга, тем точнее анализ. Этих таблиц составлено довольно большое количество, причем они могут иметь применение в любой лаборатории, т. к. точно известны условия разрядов при их составлении и эти условия м. б. совершенно точно воспроизведены. Достигается это при помощи следующего простого приема. В спектре вещества А + а% В выбираются две линии элемента А, интенсивность которых очень сильно меняется в зависимости от величины самоиндукции во вторичной цепи, именно одна дуговая (принадлежащая нейтральному атому) и одна искровая линия (принадлежащая иону). Эти две линии называются фиксирующей парой. Путем подбора величины самоиндукции линии этой пары делаются одинаковыми и составление ведется именно при этих условиях, всегда указываемых в таблицах. При таких же условиях проводится и анализ, и по осуществлению той или иной гомология, пары находится процентное содержание. Имеется несколько модификаций метода гомология, пар. Из них главнейшим является метод вспомогательного спектра, применяемый в том случае, когда элементы А и В не обладают достаточным количеством линий. В этом случае линии спектра элемента А определенным образом связываются с линиями другого, более пригодного элемента G, и роль А на чинает играть элемент G. Метод гомологии, пар разработан Герляхом и Швейтцером [3]. Он применим как к сплавам, так и к растворам. Его точность в среднем ок. ±10%.
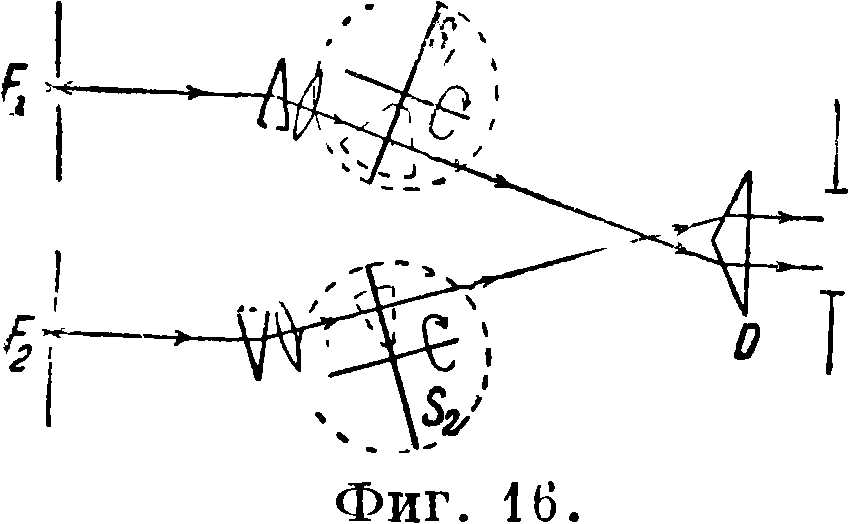
В. Методы с применением фотометрии. 1) Метод Барратга [п]. Фигура 16 дает представление о методе. F±m F2—
два искровых промежутка, при помощи которых одновременно возбуждаются спектры стандартного и анализируемого вещества. Свет проходит через 2 вращающихся сектора iS’j и S2 и при помощи призмы D образует спектры, которые расположены один над другим. Путем подбора вырезок секторов линии исследуемого элемента получают одинаковую интенсивность; концентрация определяемого элемента вычисляется из со
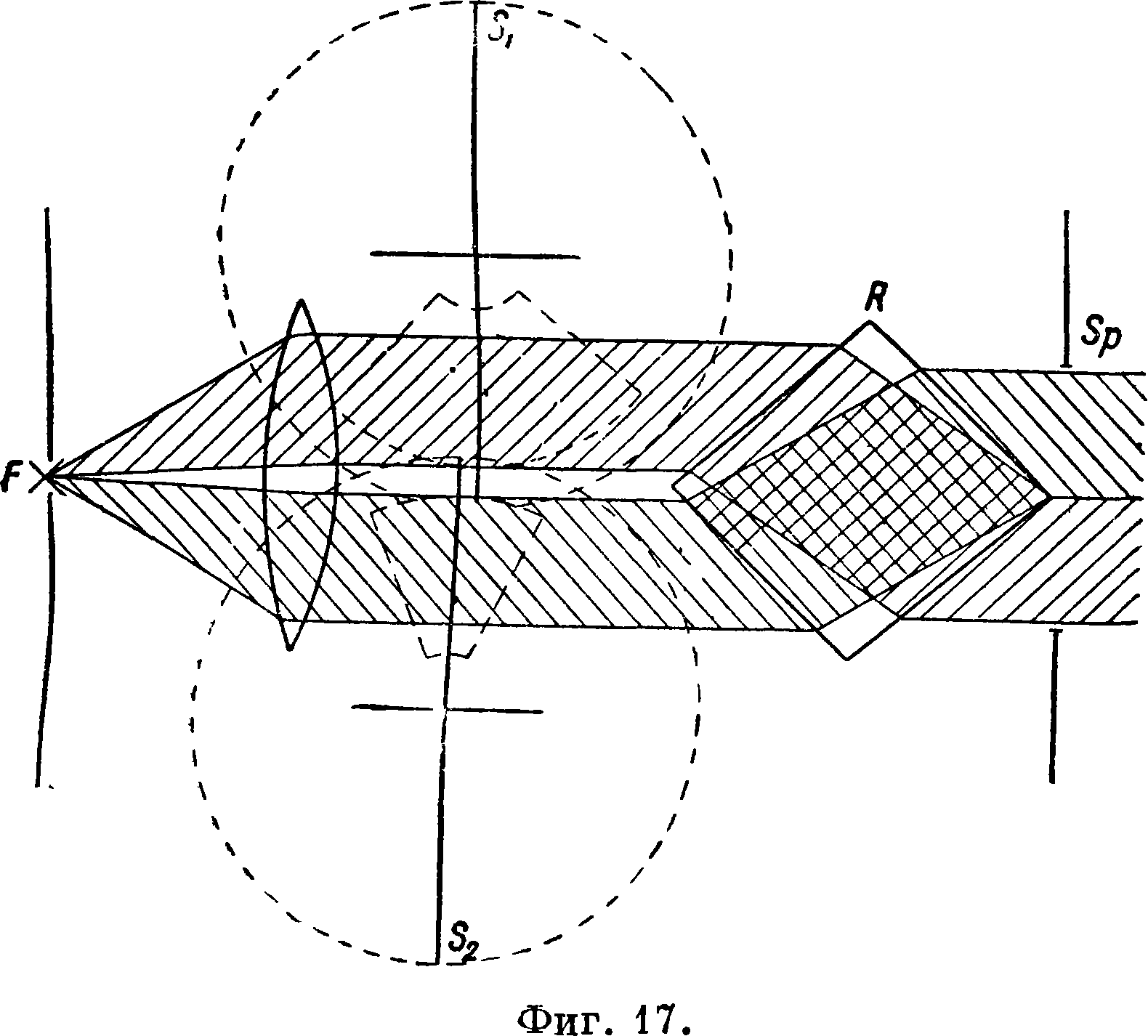
отношения величин вырезок. 2) Метод Шей-бе и Шмидта [5] является аналогичным, но с одним искровым промежутком (фигура 17). Свет от F разделяется на два пучка и проходит через секторы и *S2; при помощи ромба Гюфнера R
Фигура 18.
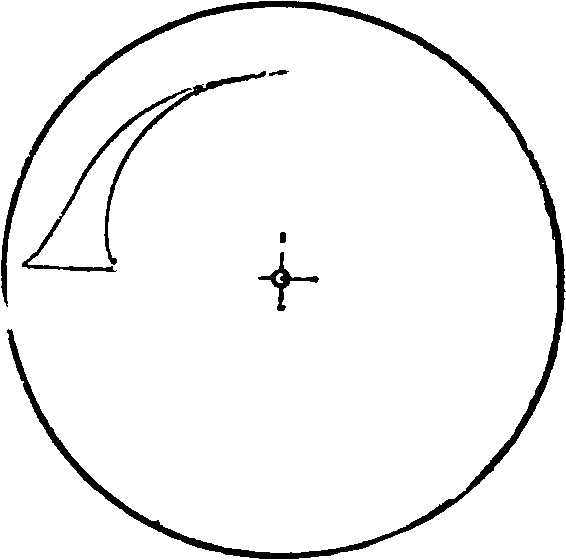
две полоски спектра получаются одна над другой; Sp — щель спектрографа. Вырезки секторов изменяются до получения равенства интенсивности линии примеси и какой-либо близлежащей линии основного вещества и по соотношению величин вырезок высчитывается %-ное содержание определяемого элемента. 3) При применении в качестве фотометра вращающегося логарифмического сектора [4U, 41] линии получают на спектрограммах клинообразный вид. Один из таких секторов и его поло жение относительно спектрографа при работе изображены на фигуре 18, а и б. Вырезка сектора подчиняется ур-ию
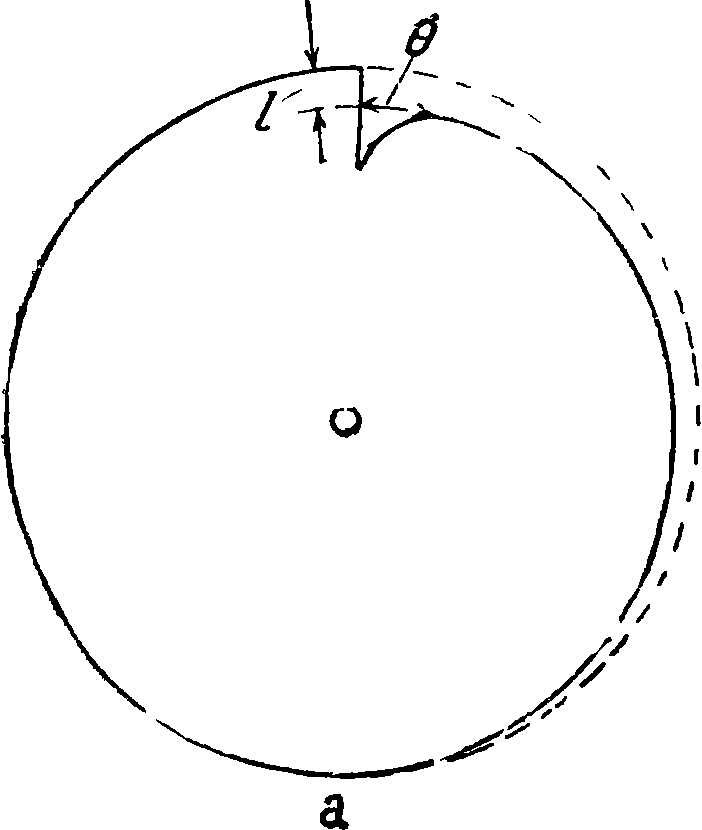
— lg0=0,3 + 0,2/,
где Θ — длина дуги в частях полной окружности, находящаяся на расстоянии измеренном в миллиметров по радиусу от его конца. Мерой интенсивности линий является их длина, т. к. с изменением концентрации элемента длина его клинообразных линий также изменяется. Предварительно по образцам с известным содержанием строится диаграмма зависимости длины какой-либо линии от %-ного содержания; при анализе на спектрограмме измеряется длина той же линии и по диаграмме находится процентное содержание. Имеется несколько различных модификаций этого метода. Следует указать на модификацию Шейбе [5], применявшего т. н. двойной логарифмич. сектор. Вид этого сектора изображен на фигуре 19. Линии исследуются затем при помощи специального аппарата. Точность, достижимая при помощи логарифмич. секторов, +10 — 15%; модификация Шейбе дает точность +5—7%. 4) Довольно часто применяется фотометрирование спектральных линий при помощи свето- и термоэлектрич. спектрофотометров самых различных конструкций [“, 5, 42, 43]. Удобными являются термоэлектрич. фотометры, выработанные специально для целей количественного анализа. Для примера на фигуре 20 приведена схема фотометра по Шейбе:
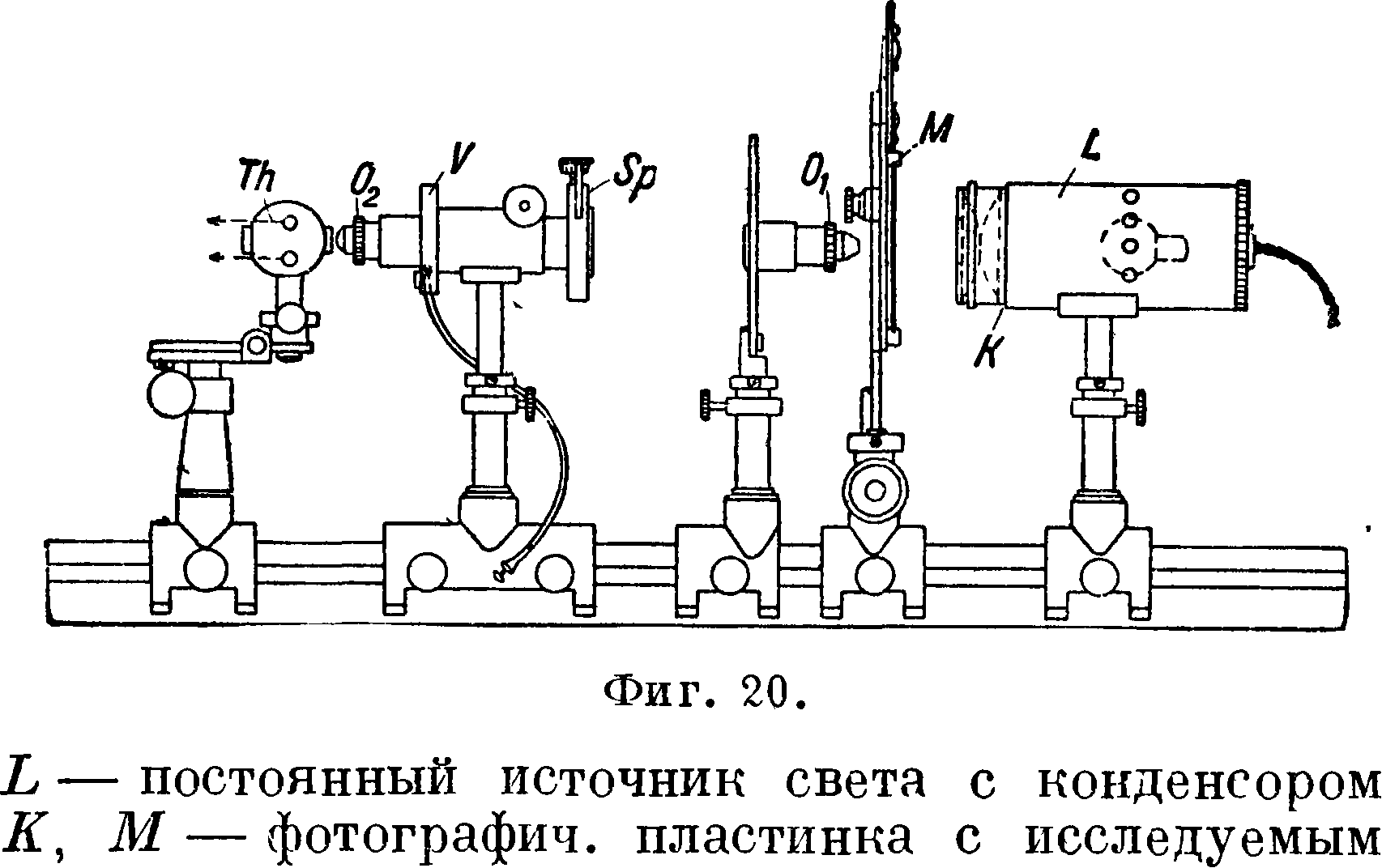
спектром, Sp — щель, Ог и 02 — объективы, V — затвор, Th — термоэлемент, κ-рый присоединяется к гальванометру. Мерой интенсивности линий является отклонение стрелки гальванометра. Реже пользуются саморегистрирующими гальванометрами, дающими запись интенсивности линий в виде кривой. Точность анализа при применении этого типа фотометрии составляет +5— 10%. При сочетании с другими методами количественного анализа точность м. б. повышена; так например, метод трех линий Шейбе и Щнеттлера [5, “], являющийся сочетанием метода гомологии, пар и фотометрии, измерений, в благоприятных случаях может дать точность+1—2%. См. Спектральный анализ и Спектрофотометрия.
Лит.: 1) Lundegardh Н., Die quantitative Spektralanalyse der Elemente, Jena, 1929; 2) L un
de g a r d h H., Die quantitative Spektralanalyse der Elemente, Teii 2, Jena, 1934; 3) G e r 1 a c h W. u.
SchweitzerE., Die chemische Emissionsspektralana-lyse, Lpz., 1930; 4) G e r 1 a c h W. u. Gerlach W., Die chemische Emissionsspektralanalyse, T. 2, Lpz., 1933 (со списком литературы); δ) Scheibe G·, Chemische Spektralanalyse, в книге: Physikalische Methoden der Analytischen Chemie, hrsg. von W. Bottger,T. l,Lpz., 1933 (со списком литературы); β) Lewis J., Spectroscopy in
Science and Industry, L., 1,933; 7) L ϋ we F., Optische Mes-sungen des Chemikers und des Mediziners, Dresden, 1933;
8) БоровикС. и Филиппов А., Спектральный анализ, «Современные физико-химические методы химического анализа», Сб. ст. под ред. С. Щукарева, вып. 1, М.—Л., 1932; 9) The Practice of Spectrum Analysis
with Hilger Instruments, L., 1935 (со списком литературы); Ю) Русанов А., «Журн. прикл. хим.», 1935, т. 8, № 3; ϋ) В а г г a 11, Поляризационный фотометр при работе с двумя искрами, Ан. П. 320136/28; 12) S с h e i-b e G., «Ztschr. f. angew. Ch.», 1931, В. 44; 13) S с h e i-be G. und Limmer G., «Metallwirtschaft», Berlin, 1932, В. 11; 14) Scheibe G. u. Limmer G., «For-schungsberichte des Gutehoffnungshiitte-Konzerns», 1932, 2; ΐδ) Occhialini H., «Atti della Academia dei Lin-cei», Roma, 1929, s. 6, v. 9, 1931, v. 13; i«) Corsi A., «Nuova Cimento», 1929, 6;17) Lockyer J. a. Roberts W., «Proceedings of the Roy. Soc. of London», 1873, v. 21; 18) Lundegardh H., «Ztschr. f. Physik», B., 1930, 66; Ю) Ландсберг Г., Мандельштам С., Райский С., «Журн. технической оизики» (1933), т. 3;#вып. 5; 20) Russanow A., «Z. f. analyt. Chem.», Mch., 1935, В. 38; 2i) p о 11 о k J. a. Leonard A., «Proceedings of the Roy. Soc. of Dublin», 1907, 11; 22) Leonard
A., ibid., 1908, 11; 23) p 0 1 1 о k J., ibid., 1909, 11; *4) Leonard A. a. Whelan P., ibid., 1918, 15; 2δ) Hartley W., «Journ. of the Chem. Soc. of London», 1884, y. 41; 26) De G r a m ο n t A., «Bui. de la So-ci6te chimique de France», P., 1923, s. 4, t. 33; 27) q e i 1-m a η n W. u. В r ii nger K., «Ztschr. f. anorg. Chem.», Lpz., 1931, B. 196; 28) Wach6 X., «CR», 1923, t. 177; 29) F e s e f e 1 d t H., «Ztschr. f. physikal. Chemie», Lpz., 1929, B. 140; 30) Kohlrausch F., Lehrbuch d. pra-ktischen Physik, Mch.—B., 16 Aufl., 1930; 31) N e g r e-s с о T., «CR», 1927, t. 185; 32) Laszlo H., «Ind. Eng. Chem.», 1927, v. 19; зз) Reis A., «Naturwissenschaft», В., 1926, В. 14; 34) N i t c h i e C., «ind. Eng. Chem.», 1929, 1; 3δ) Green J., «Journal of the Soc. of Chem. Ind.», 1927, v. 46; 36) К e 1 1 e r m a η n K. u. S c h 1 i e s s m a η n O., «Metallborse», B., 1927, B. 17; 37) Thurnwald H. u. H u t t i g G., «Ztschr. f. anal. Chem.», Mch., 1929,
B. 76 и 77; 38) Пустовалов Л., К методике спектральных исследований минеральных тел, «Труды ин-та мин. и петрографии», М., 1926, вып. 4; 39) Метод и таблицы гомологических пар 3—5; 40) Лизунов Н., «Заводская лаборатория», 1935, δ; 41) Twyman F. а. H i t с h e n C., «Proc. of the Roy. Soc. of London», 1931, s. A., γ. 133; 42) Ruthardt W., «Ztschr. f. anorg. Chem.», Lpz., 1931, B. 195; 43) Urbain P., «Bulletin de la Soc. chim. de France», P., 1930, s. 4. t. 47; 44) Scheibe G. u. Schnettler O., «Naturwissenschaft», 1930, B. 18, 1931, B. 19 (Таблицы длин волны); 45) к ° y s er H. Tabelle der Hauptlinien der Liniensp^ktra a le" Elemente, B., 1926; 46) e x n e r F. u. Haschek E., Die Spektren der Elemente bei normalem Druck, W., 1911 /12; 47) e d e r J. u. V a 1 e n t a E., Atlas tYpischer Spektren, W., 1924; 48) Smith D., Visual Lines for Spectrum Analysis, L., 1928; 49) Twyman F. a. Smith D., Wavelength Tables for Spectrum Analysis, L., 1931 (со списком всех работ Грамона);
50) L б w e F., Atlasd. letztenLinien, Dresden—Lpz., 1928;
51) D e G r a m ο n t A., «CR», 1920, t. 171. H. Лизунов.